Wuming Xie, Baoqiong Liao, Mei Shuai, Rutian Liu, Min Hong, Shuwen He
{"title":"与神经发育障碍相关的SYNGAP1新的从头内含子变异。","authors":"Wuming Xie, Baoqiong Liao, Mei Shuai, Rutian Liu, Min Hong, Shuwen He","doi":"10.1002/mgg3.70066","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>SYNGAP1 encodes a Ras/Rap GTPase-activating protein that is predominantly expressed in the brain with the functional roles in regulating synaptic plasticity, spine morphogenesis, and cognition function. Pathogenic variants in SYNGAP1 have been associated with a spectrum of neurodevelopmental disorders characterized by developmental delays, intellectual disabilities, epilepsy, hypotonia, and the features of autism spectrum disorder. The aim of this study was to identify a novel SYNGAP1 gene variant linked to neurodevelopmental disorders and to evaluate the pathogenicity of the detected variant.</p><p><strong>Methods: </strong>A novel de novo intronic variant in SYNGAP1 was identified by Whole exome sequencing (WES) and confirmed by Sanger sequencing. Minigene assays were conducted to assess whether the intronic variant in SYNGAP1 influenced the normal splicing of mRNA.</p><p><strong>Results: </strong>A novel de novo intronic variant in SYNGAP1 (c.3582+2T>G) was indentified with clinical features suggestive of neurodevelopmental related disorders. Minigene splicing analysis demonstrated that this noncanonical splice site variant led to the activation of a cryptic acceptor splice site. Consequently, 101 base pairs of intron 16 were aberrantly retained in the mRNA, leading to a frameshift. This frameshift resulted in the introduction of a premature stop codon (TGA) in the coding sequence and the production of a truncated SYNGAP1 protein, potentially leding to loss of function and subsequent disruption of its biological roles.</p><p><strong>Conclusion: </strong>Our findings highlight the significance of de novo pathogenic SYNGAP1 variants at the intron 16/exon 17 junction in the SYNGAP1-related neurodevelopmental disorders, providing novel insights into the genetic basis and diagnosis of these disabilities.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 2","pages":"e70066"},"PeriodicalIF":1.6000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11775916/pdf/","citationCount":"0","resultStr":"{\"title\":\"Novel De Novo Intronic Variant of SYNGAP1 Associated With the Neurodevelopmental Disorders.\",\"authors\":\"Wuming Xie, Baoqiong Liao, Mei Shuai, Rutian Liu, Min Hong, Shuwen He\",\"doi\":\"10.1002/mgg3.70066\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>SYNGAP1 encodes a Ras/Rap GTPase-activating protein that is predominantly expressed in the brain with the functional roles in regulating synaptic plasticity, spine morphogenesis, and cognition function. Pathogenic variants in SYNGAP1 have been associated with a spectrum of neurodevelopmental disorders characterized by developmental delays, intellectual disabilities, epilepsy, hypotonia, and the features of autism spectrum disorder. The aim of this study was to identify a novel SYNGAP1 gene variant linked to neurodevelopmental disorders and to evaluate the pathogenicity of the detected variant.</p><p><strong>Methods: </strong>A novel de novo intronic variant in SYNGAP1 was identified by Whole exome sequencing (WES) and confirmed by Sanger sequencing. Minigene assays were conducted to assess whether the intronic variant in SYNGAP1 influenced the normal splicing of mRNA.</p><p><strong>Results: </strong>A novel de novo intronic variant in SYNGAP1 (c.3582+2T>G) was indentified with clinical features suggestive of neurodevelopmental related disorders. Minigene splicing analysis demonstrated that this noncanonical splice site variant led to the activation of a cryptic acceptor splice site. Consequently, 101 base pairs of intron 16 were aberrantly retained in the mRNA, leading to a frameshift. This frameshift resulted in the introduction of a premature stop codon (TGA) in the coding sequence and the production of a truncated SYNGAP1 protein, potentially leding to loss of function and subsequent disruption of its biological roles.</p><p><strong>Conclusion: </strong>Our findings highlight the significance of de novo pathogenic SYNGAP1 variants at the intron 16/exon 17 junction in the SYNGAP1-related neurodevelopmental disorders, providing novel insights into the genetic basis and diagnosis of these disabilities.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"13 2\",\"pages\":\"e70066\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2025-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11775916/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.70066\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70066","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Novel De Novo Intronic Variant of SYNGAP1 Associated With the Neurodevelopmental Disorders.
Background: SYNGAP1 encodes a Ras/Rap GTPase-activating protein that is predominantly expressed in the brain with the functional roles in regulating synaptic plasticity, spine morphogenesis, and cognition function. Pathogenic variants in SYNGAP1 have been associated with a spectrum of neurodevelopmental disorders characterized by developmental delays, intellectual disabilities, epilepsy, hypotonia, and the features of autism spectrum disorder. The aim of this study was to identify a novel SYNGAP1 gene variant linked to neurodevelopmental disorders and to evaluate the pathogenicity of the detected variant.
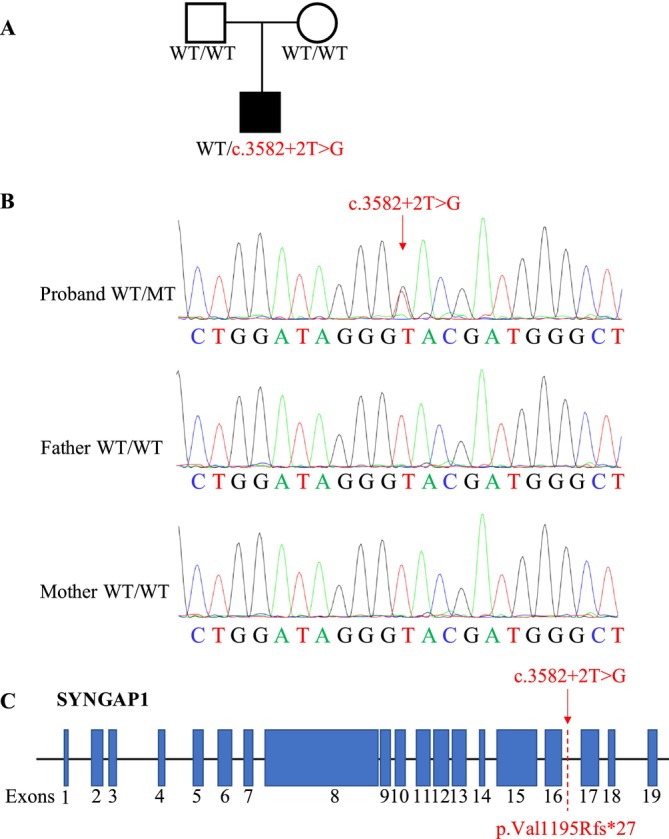
Methods: A novel de novo intronic variant in SYNGAP1 was identified by Whole exome sequencing (WES) and confirmed by Sanger sequencing. Minigene assays were conducted to assess whether the intronic variant in SYNGAP1 influenced the normal splicing of mRNA.
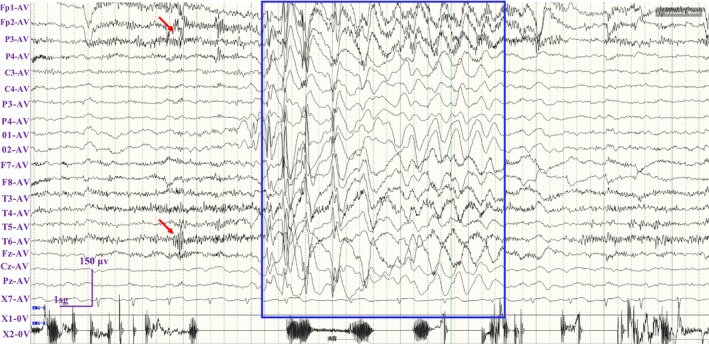
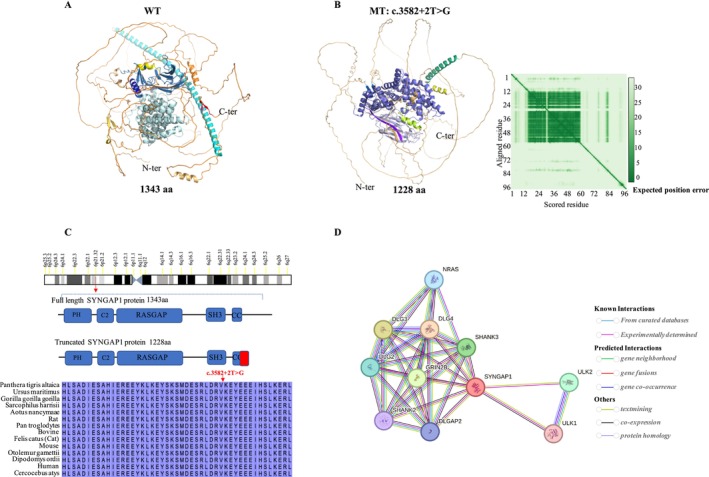
Results: A novel de novo intronic variant in SYNGAP1 (c.3582+2T>G) was indentified with clinical features suggestive of neurodevelopmental related disorders. Minigene splicing analysis demonstrated that this noncanonical splice site variant led to the activation of a cryptic acceptor splice site. Consequently, 101 base pairs of intron 16 were aberrantly retained in the mRNA, leading to a frameshift. This frameshift resulted in the introduction of a premature stop codon (TGA) in the coding sequence and the production of a truncated SYNGAP1 protein, potentially leding to loss of function and subsequent disruption of its biological roles.
Conclusion: Our findings highlight the significance of de novo pathogenic SYNGAP1 variants at the intron 16/exon 17 junction in the SYNGAP1-related neurodevelopmental disorders, providing novel insights into the genetic basis and diagnosis of these disabilities.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


