Regina de Miguel Ibañez, Pattsy Etual Espinosa Cárdenas, Manuel Ramón García Sáenz, Bayron A Sandoval Bonilla
{"title":"神经结节病的全垂体功能减退:利妥昔单抗成功治疗垂体-下丘脑受累。","authors":"Regina de Miguel Ibañez, Pattsy Etual Espinosa Cárdenas, Manuel Ramón García Sáenz, Bayron A Sandoval Bonilla","doi":"10.1210/jcemcr/luaf014","DOIUrl":null,"url":null,"abstract":"<p><p>Neurosarcoidosis (NS) is a rare form of sarcoidosis, with isolated hypothalamic-pituitary involvement being exceptionally uncommon. We report a 20-year-old woman presenting with polyuria, galactorrhea, amenorrhea, and substantial weight loss. Hormonal evaluation revealed hypopituitarism with arginine-vasopressin deficiency and hyperprolactinemia. Magnetic resonance imaging demonstrated pituitary stalk thickening and suprasellar extension, initially suggestive of hypophysitis. High-dose glucocorticoid therapy resulted in partial regression of the pituitary lesion but persistence of suprasellar involvement, prompting a transcranial stereotactic biopsy. Histopathological analysis confirmed isolated NS with noncaseating granulomas. The patient was treated with rituximab after partial response to glucocorticoids, achieving significant clinical and radiological improvement, although hormonal axis recovery was not observed. Hormone replacement therapy remains necessary. The case met the criteria for definitive type b NS, as no extraneural involvement was identified. This case underscores the diagnostic challenges of isolated NS and highlights the importance of considering histopathological confirmation in patients without systemic manifestations to guide treatment. Glucocorticoids are first-line therapy, but rituximab may be effective as a second-line option for refractory cases. Early diagnosis and tailored therapy are essential to improving outcomes in this rare and challenging condition.</p>","PeriodicalId":73540,"journal":{"name":"JCEM case reports","volume":"3 2","pages":"luaf014"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11758143/pdf/","citationCount":"0","resultStr":"{\"title\":\"Panhypopituitarism in Neurosarcoidosis: Pituitary-Hypothalamic Involvement Successfully Managed With Rituximab.\",\"authors\":\"Regina de Miguel Ibañez, Pattsy Etual Espinosa Cárdenas, Manuel Ramón García Sáenz, Bayron A Sandoval Bonilla\",\"doi\":\"10.1210/jcemcr/luaf014\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Neurosarcoidosis (NS) is a rare form of sarcoidosis, with isolated hypothalamic-pituitary involvement being exceptionally uncommon. We report a 20-year-old woman presenting with polyuria, galactorrhea, amenorrhea, and substantial weight loss. Hormonal evaluation revealed hypopituitarism with arginine-vasopressin deficiency and hyperprolactinemia. Magnetic resonance imaging demonstrated pituitary stalk thickening and suprasellar extension, initially suggestive of hypophysitis. High-dose glucocorticoid therapy resulted in partial regression of the pituitary lesion but persistence of suprasellar involvement, prompting a transcranial stereotactic biopsy. Histopathological analysis confirmed isolated NS with noncaseating granulomas. The patient was treated with rituximab after partial response to glucocorticoids, achieving significant clinical and radiological improvement, although hormonal axis recovery was not observed. Hormone replacement therapy remains necessary. The case met the criteria for definitive type b NS, as no extraneural involvement was identified. This case underscores the diagnostic challenges of isolated NS and highlights the importance of considering histopathological confirmation in patients without systemic manifestations to guide treatment. Glucocorticoids are first-line therapy, but rituximab may be effective as a second-line option for refractory cases. Early diagnosis and tailored therapy are essential to improving outcomes in this rare and challenging condition.</p>\",\"PeriodicalId\":73540,\"journal\":{\"name\":\"JCEM case reports\",\"volume\":\"3 2\",\"pages\":\"luaf014\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-01-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11758143/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JCEM case reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1210/jcemcr/luaf014\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/2/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JCEM case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jcemcr/luaf014","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
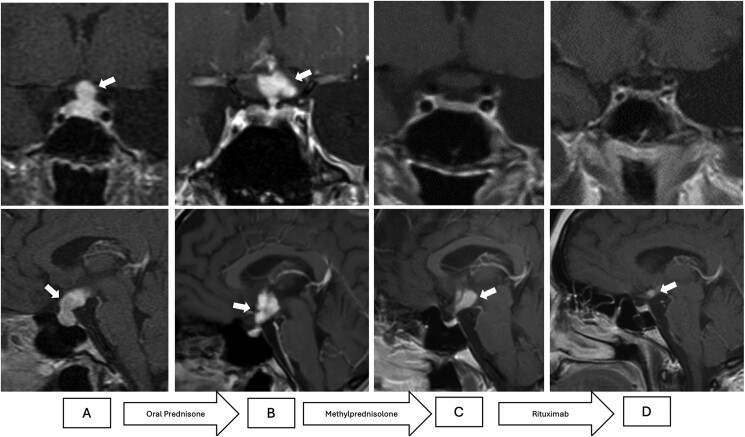
神经肉芽肿病(NS)是一种罕见的肉芽肿病,孤立的下丘脑-垂体受累异常罕见。我们报告了一名 20 岁女性的病例,她出现多尿、半乳溢乳、闭经和体重大幅下降。激素评估显示她患有垂体功能减退症,伴有精氨酸-血管加压素缺乏症和高泌乳素血症。磁共振成像显示垂体柄增粗并向星状上部扩展,初步提示为垂体功能减退症。大剂量糖皮质激素治疗导致垂体病变部分消退,但星状上部受累持续存在,促使患者进行经颅立体定向活检。组织病理学分析证实,孤立性NS伴有非溃疡性肉芽肿。患者对糖皮质激素部分反应后接受了利妥昔单抗治疗,临床和放射学症状明显改善,但激素轴仍未恢复。仍需进行激素替代治疗。该病例符合确定性 b 型 NS 的标准,因为没有发现硬膜外受累。该病例凸显了孤立性 NS 在诊断方面的挑战,并强调了考虑对无全身表现的患者进行组织病理学确诊以指导治疗的重要性。糖皮质激素是一线疗法,但利妥昔单抗作为二线疗法可能对难治性病例有效。早期诊断和有针对性的治疗对于改善这种罕见且具有挑战性的疾病的预后至关重要。
Panhypopituitarism in Neurosarcoidosis: Pituitary-Hypothalamic Involvement Successfully Managed With Rituximab.
Neurosarcoidosis (NS) is a rare form of sarcoidosis, with isolated hypothalamic-pituitary involvement being exceptionally uncommon. We report a 20-year-old woman presenting with polyuria, galactorrhea, amenorrhea, and substantial weight loss. Hormonal evaluation revealed hypopituitarism with arginine-vasopressin deficiency and hyperprolactinemia. Magnetic resonance imaging demonstrated pituitary stalk thickening and suprasellar extension, initially suggestive of hypophysitis. High-dose glucocorticoid therapy resulted in partial regression of the pituitary lesion but persistence of suprasellar involvement, prompting a transcranial stereotactic biopsy. Histopathological analysis confirmed isolated NS with noncaseating granulomas. The patient was treated with rituximab after partial response to glucocorticoids, achieving significant clinical and radiological improvement, although hormonal axis recovery was not observed. Hormone replacement therapy remains necessary. The case met the criteria for definitive type b NS, as no extraneural involvement was identified. This case underscores the diagnostic challenges of isolated NS and highlights the importance of considering histopathological confirmation in patients without systemic manifestations to guide treatment. Glucocorticoids are first-line therapy, but rituximab may be effective as a second-line option for refractory cases. Early diagnosis and tailored therapy are essential to improving outcomes in this rare and challenging condition.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


