乙苯在H-BEA分子筛内单分子异构化的第一性原理研究
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
为了破译石化工业中涉及乙苯的反应网络,利用密度泛函理论研究了乙苯单分子异构化制二甲苯的过程。通过从头算分子动力学模拟和元动力学的结合,确定了表征乙烯/亚甲基转移、Buchner和逆Buchner环扩张、分子内氢化物转移、阳离子双环[4.1.0]烃去质子化和去甲二烯质子化八个基本步骤的自由能图。发现Buchner和反Buchner环展开是限速步骤,其自由能垒最高为205 kJ/mol。而通过反向Friedel-Crafts烷基化步骤对阳离子二芳基中间体的破坏在动力学上是可比的。进一步的电荷分析表明,Buchner和反Buchner环扩展步骤的最高自由能势垒可归因于两个强正电荷原子群之间的电子转移。这项全面的计算研究为乙苯异构化过程的复杂细节提供了有价值的见解,揭示了石化工业中这一重要反应的潜在机制。本文章由计算机程序翻译,如有差异,请以英文原文为准。
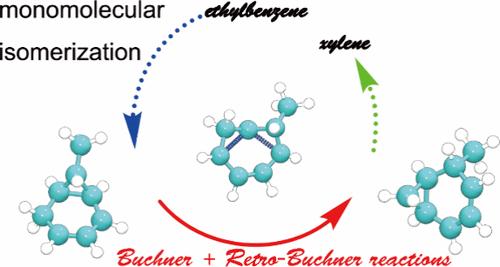
First-Principles Study on the Monomolecular Isomerization of Ethylbenzene within H-BEA Zeolite
To decipher the reaction network involving ethylbenzene in the petrochemical industry, the monomolecular isomerization process of ethylbenzene to xylene has been studied using density functional theory. With a combination of ab initio molecular dynamics simulations and metadynamics, free energy landscapes characterizing eight elementary steps, viz., ethylidene/methylene transfer, Buchner and retro-Buchner ring expansion, intramolecular hydride transfer, deprotonation of cationic bicyclo[4.1.0] hydrocarbon, and protonation of norcaradiene, have been determined. The Buchner and retro-Buchner ring expansions are found to be the rate-limiting step with the highest free energy barrier of 205 kJ/mol. While the destruction of the cationic diaryl intermediate via the reverse Friedel–Crafts alkylation step is kinetically comparable. Further charge analysis reveals that the highest free energy barrier for the Buchner and retro-Buchner ring expansion step can be attributed to the electron transfer between two strongly positively charged atom groups. This comprehensive computational study offers valuable insights into the intricate details of the ethylbenzene isomerization process, shedding light on the underlying mechanisms of this important reaction in the petrochemical industry.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


