{"title":"Sn的可用原子间电位对其二维同素异形体的适用性","authors":"Marcin Maździarz","doi":"10.1002/jcc.70032","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The suitability of a range of interatomic potentials for elemental tin was evaluated in order to identify an appropriate potential for modeling the stanene (2D tin) allotropes. Structural and mechanical features of the flat (F), low-buckled (LB), high-buckled (HB), full dumbbell (FD), trigonal dumbbell (TD), honeycomb dumbbell (HD), and large honeycomb dumbbell (LHD) monolayer tin (stanene) phases, were gained by means of the density functional theory (DFT) and molecular statics (MS) calculations with ten different Tersoff, modified embedded atom method (MEAM), and machine-learning-based (ML-IAP) interatomic potentials. A systematic quantitative comparison and discussion of the results is reported.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 2","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Suitability of Available Interatomic Potentials for Sn to Model Its 2D Allotropes\",\"authors\":\"Marcin Maździarz\",\"doi\":\"10.1002/jcc.70032\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>The suitability of a range of interatomic potentials for elemental tin was evaluated in order to identify an appropriate potential for modeling the stanene (2D tin) allotropes. Structural and mechanical features of the flat (F), low-buckled (LB), high-buckled (HB), full dumbbell (FD), trigonal dumbbell (TD), honeycomb dumbbell (HD), and large honeycomb dumbbell (LHD) monolayer tin (stanene) phases, were gained by means of the density functional theory (DFT) and molecular statics (MS) calculations with ten different Tersoff, modified embedded atom method (MEAM), and machine-learning-based (ML-IAP) interatomic potentials. A systematic quantitative comparison and discussion of the results is reported.</p>\\n </div>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 2\",\"pages\":\"\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2025-01-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70032\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70032","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Suitability of Available Interatomic Potentials for Sn to Model Its 2D Allotropes
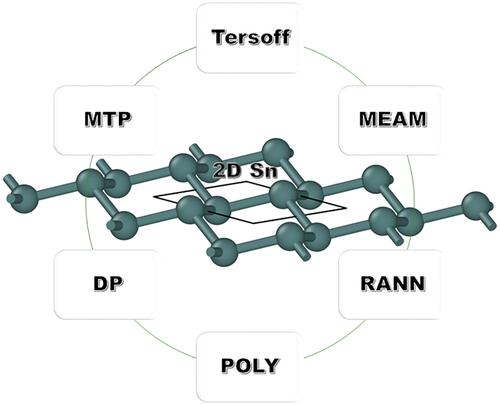
The suitability of a range of interatomic potentials for elemental tin was evaluated in order to identify an appropriate potential for modeling the stanene (2D tin) allotropes. Structural and mechanical features of the flat (F), low-buckled (LB), high-buckled (HB), full dumbbell (FD), trigonal dumbbell (TD), honeycomb dumbbell (HD), and large honeycomb dumbbell (LHD) monolayer tin (stanene) phases, were gained by means of the density functional theory (DFT) and molecular statics (MS) calculations with ten different Tersoff, modified embedded atom method (MEAM), and machine-learning-based (ML-IAP) interatomic potentials. A systematic quantitative comparison and discussion of the results is reported.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


