生成式人工智能设计的卤代烷烃脱卤酶变体的生化和计算表征:加速SN2步骤
IF 15.6
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
基于天然蛋白质序列训练的生成式人工智能(AI)模型已被用于设计功能酶。然而,它们预测酶催化中单个反应步骤的能力尚不清楚,这限制了序列信息在酶工程中的潜在应用。在这项研究中,我们证明了序列信息可以使用生成最大熵(MaxEnt)模型预测卤代烷烃脱卤酶SN2步骤的速率。然后,我们利用该模型设计了卤烷脱卤酶的低阶蛋白变体。动力学测量证实了蛋白质变体的成功设计,这些变体在整个反应中,特别是在SN2步骤中,提高了催化活性,高于野生型。在模拟方面,我们使用经验价键(EVB)和元动力学模拟对SN2步骤的这些设计提供了分子见解。EVB计算结果表明,激活势垒与实验反应速率一致,同时考察了氨基酸取代对静电对激活势垒的影响、水渗透的后果以及基态不稳定/稳定的程度。元动力学模拟强调了酶催化中底物定位的重要性。总体而言,我们的人工智能指导方法成功地设计了一个SN2步骤速度比野生型酶更快的变体,尽管卤代烷烃脱卤酶通过自然进化得到了广泛的优化。本文章由计算机程序翻译,如有差异,请以英文原文为准。
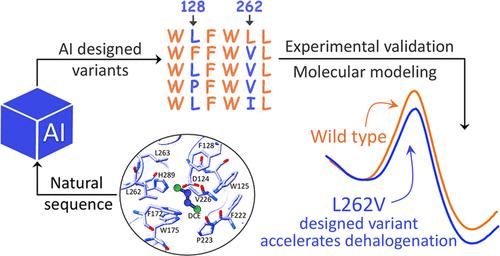
Biochemical and Computational Characterization of Haloalkane Dehalogenase Variants Designed by Generative AI: Accelerating the SN2 Step
Generative artificial intelligence (AI) models trained on natural protein sequences have been used to design functional enzymes. However, their ability to predict individual reaction steps in enzyme catalysis remains unclear, limiting the potential use of sequence information for enzyme engineering. In this study, we demonstrated that sequence information can predict the rate of the SN2 step of a haloalkane dehalogenase using a generative maximum-entropy (MaxEnt) model. We then designed lower-order protein variants of haloalkane dehalogenase using the model. Kinetic measurements confirmed the successful design of protein variants that enhance catalytic activity, above that of the wild type, in the overall reaction and in particular in the SN2 step. On the simulation side, we provided molecular insights into these designs for the SN2 step using the empirical valence bond (EVB) and metadynamics simulations. The EVB calculations showed activation barriers consistent with experimental reaction rates, while examining the effect of amino acid replacements on the electrostatic effect on the activation barrier and the consequence of water penetration, as well as the extent of ground state destabilization/stabilization. Metadynamics simulations emphasize the importance of the substrate positioning in enzyme catalysis. Overall, our AI-guided approach successfully enabled the design of a variant with a faster rate for the SN2 step than the wild-type enzyme, despite haloalkane dehalogenase being extensively optimized through natural evolution.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


