Daniel Medina-Neira, Giancarlo Alvarado-Gamarra, Brenda Huamaní-Condori, Nelson Purizaca-Rosillo, Noé Atamari-Anahui, Erick Matos-Villena, Raquel Garces-Ghilardi, Matilde Estupiñan-Vigil
{"title":"嗜血球性淋巴组织细胞增多症是TERC变异端粒生物学障碍患儿骨髓衰竭的初始表现。","authors":"Daniel Medina-Neira, Giancarlo Alvarado-Gamarra, Brenda Huamaní-Condori, Nelson Purizaca-Rosillo, Noé Atamari-Anahui, Erick Matos-Villena, Raquel Garces-Ghilardi, Matilde Estupiñan-Vigil","doi":"10.1177/26330040241311621","DOIUrl":null,"url":null,"abstract":"<p><p>Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening systemic hyperinflammatory syndrome, rarely associated with bone marrow failure (BMF). Telomere biology disorders (TBD) are caused by inherited defects in telomerase processes and can have heterogeneous presentations including idiopathic pulmonary fibrosis, cirrhosis, and BMF. We report a case of a 10-year-old male from Lima, Peru, who presented with HLH as the initial manifestation of a TBD. He experienced fever, gastrointestinal symptoms, and mucocutaneous involvement. Initial laboratory analyses revealed pancytopenia and elevated inflammatory markers. Despite symptomatic and antibiotic treatment, his clinical condition persisted leading to a suspicion of Kawasaki disease and, subsequently, HLH. Immunomodulatory treatment was initiated with a good clinical response. Bone marrow aspiration revealed severe hypocellular bone marrow and cytophagocytosis. Genetic studies identified a pathogenic variant in the <i>TERC</i> gene (n.110_113del), which was also found in the patient's mother and brother. HLH as the initial manifestation of BMF is rare. This case highlights the importance of considering TBD in children with BMF of unclear etiology and the value of genetic testing in such cases.</p>","PeriodicalId":75218,"journal":{"name":"Therapeutic advances in rare disease","volume":"6 ","pages":"26330040241311621"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11705338/pdf/","citationCount":"0","resultStr":"{\"title\":\"Hemophagocytic lymphohistiocytosis as the initial manifestation of bone marrow failure in a child with a TERC variant telomere biology disorder.\",\"authors\":\"Daniel Medina-Neira, Giancarlo Alvarado-Gamarra, Brenda Huamaní-Condori, Nelson Purizaca-Rosillo, Noé Atamari-Anahui, Erick Matos-Villena, Raquel Garces-Ghilardi, Matilde Estupiñan-Vigil\",\"doi\":\"10.1177/26330040241311621\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening systemic hyperinflammatory syndrome, rarely associated with bone marrow failure (BMF). Telomere biology disorders (TBD) are caused by inherited defects in telomerase processes and can have heterogeneous presentations including idiopathic pulmonary fibrosis, cirrhosis, and BMF. We report a case of a 10-year-old male from Lima, Peru, who presented with HLH as the initial manifestation of a TBD. He experienced fever, gastrointestinal symptoms, and mucocutaneous involvement. Initial laboratory analyses revealed pancytopenia and elevated inflammatory markers. Despite symptomatic and antibiotic treatment, his clinical condition persisted leading to a suspicion of Kawasaki disease and, subsequently, HLH. Immunomodulatory treatment was initiated with a good clinical response. Bone marrow aspiration revealed severe hypocellular bone marrow and cytophagocytosis. Genetic studies identified a pathogenic variant in the <i>TERC</i> gene (n.110_113del), which was also found in the patient's mother and brother. HLH as the initial manifestation of BMF is rare. This case highlights the importance of considering TBD in children with BMF of unclear etiology and the value of genetic testing in such cases.</p>\",\"PeriodicalId\":75218,\"journal\":{\"name\":\"Therapeutic advances in rare disease\",\"volume\":\"6 \",\"pages\":\"26330040241311621\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-01-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11705338/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Therapeutic advances in rare disease\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/26330040241311621\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Therapeutic advances in rare disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/26330040241311621","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Hemophagocytic lymphohistiocytosis as the initial manifestation of bone marrow failure in a child with a TERC variant telomere biology disorder.
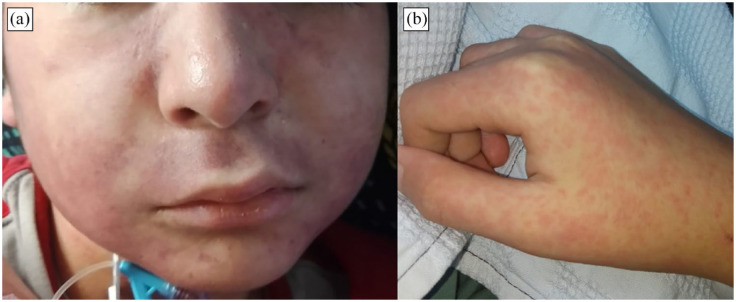
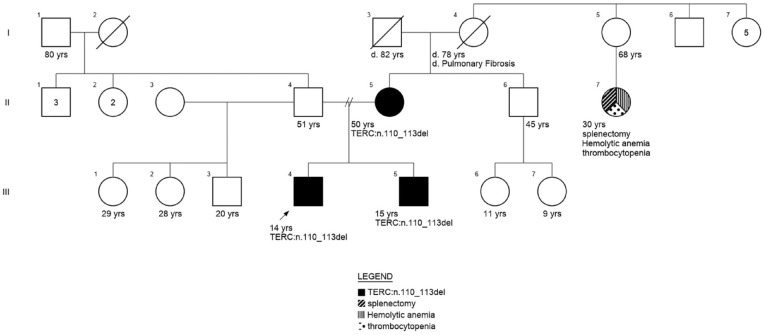
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening systemic hyperinflammatory syndrome, rarely associated with bone marrow failure (BMF). Telomere biology disorders (TBD) are caused by inherited defects in telomerase processes and can have heterogeneous presentations including idiopathic pulmonary fibrosis, cirrhosis, and BMF. We report a case of a 10-year-old male from Lima, Peru, who presented with HLH as the initial manifestation of a TBD. He experienced fever, gastrointestinal symptoms, and mucocutaneous involvement. Initial laboratory analyses revealed pancytopenia and elevated inflammatory markers. Despite symptomatic and antibiotic treatment, his clinical condition persisted leading to a suspicion of Kawasaki disease and, subsequently, HLH. Immunomodulatory treatment was initiated with a good clinical response. Bone marrow aspiration revealed severe hypocellular bone marrow and cytophagocytosis. Genetic studies identified a pathogenic variant in the TERC gene (n.110_113del), which was also found in the patient's mother and brother. HLH as the initial manifestation of BMF is rare. This case highlights the importance of considering TBD in children with BMF of unclear etiology and the value of genetic testing in such cases.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


