Hany Ghazal, El-Sayed A El-Absawy, Waleed Ead, Mohamed E Hasan
{"title":"机器学习引导的差异基因表达分析确定了三阴性乳腺癌中高度连接的七个基因簇。","authors":"Hany Ghazal, El-Sayed A El-Absawy, Waleed Ead, Mohamed E Hasan","doi":"10.37796/2211-8039.1467","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>One of the most challenging cancers is triple-negative breast cancer, which is subdivided into many molecular subtypes. Due to the high degree of heterogeneity, the role of precision medicine remains challenging. With the use of machine learning (ML)-guided gene selection, the differential gene expression analysis can be optimized, and eventually, the process of precision medicine can see great advancement through biomarker discovery.</p><p><strong>Purpose: </strong>Enhancing precision medicine in the oncology field by identification of the most representative differentially-expressed genes to be used as biomarkers or as novel drug targets.</p><p><strong>Methods: </strong>By utilizing data from the Gene Expression Omnibus (GEO) repository and The Cancer Genome Atlas (TCGA), we identified the differentially expressed genes using the linear model for microarray analysis (LIMMA) and edgeR algorithms, and applied ML-based feature selection using several algorithms.</p><p><strong>Results: </strong>A total of 27 genes were selected by merging features identified with both LIMMA and ML-based feature selection methods. The models with the highest area under the curve (AUC) are CatBoost, Extreme Gradient Boosting (XGBoost), Random Forest, and Multi-Layer Perceptron classifiers. ESR1, FOXA1, GATA3, XBP1, GREB1, AR, and AGR2 were identified as hub genes in a highly interconnected cluster.</p><p><strong>Conclusion: </strong>ML-based gene selection shows a great impact on the identification of hub genes. The ML models built can improve precision oncology in diagnosis and prognosis. The identified hub genes can serve as biomarkers and warrant further research for potential drug target development.</p>","PeriodicalId":51650,"journal":{"name":"BioMedicine-Taiwan","volume":"14 4","pages":"15-35"},"PeriodicalIF":2.5000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11703398/pdf/","citationCount":"0","resultStr":"{\"title\":\"Machine learning-guided differential gene expression analysis identifies a highly-connected seven-gene cluster in triple-negative breast cancer.\",\"authors\":\"Hany Ghazal, El-Sayed A El-Absawy, Waleed Ead, Mohamed E Hasan\",\"doi\":\"10.37796/2211-8039.1467\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>One of the most challenging cancers is triple-negative breast cancer, which is subdivided into many molecular subtypes. Due to the high degree of heterogeneity, the role of precision medicine remains challenging. With the use of machine learning (ML)-guided gene selection, the differential gene expression analysis can be optimized, and eventually, the process of precision medicine can see great advancement through biomarker discovery.</p><p><strong>Purpose: </strong>Enhancing precision medicine in the oncology field by identification of the most representative differentially-expressed genes to be used as biomarkers or as novel drug targets.</p><p><strong>Methods: </strong>By utilizing data from the Gene Expression Omnibus (GEO) repository and The Cancer Genome Atlas (TCGA), we identified the differentially expressed genes using the linear model for microarray analysis (LIMMA) and edgeR algorithms, and applied ML-based feature selection using several algorithms.</p><p><strong>Results: </strong>A total of 27 genes were selected by merging features identified with both LIMMA and ML-based feature selection methods. The models with the highest area under the curve (AUC) are CatBoost, Extreme Gradient Boosting (XGBoost), Random Forest, and Multi-Layer Perceptron classifiers. ESR1, FOXA1, GATA3, XBP1, GREB1, AR, and AGR2 were identified as hub genes in a highly interconnected cluster.</p><p><strong>Conclusion: </strong>ML-based gene selection shows a great impact on the identification of hub genes. The ML models built can improve precision oncology in diagnosis and prognosis. The identified hub genes can serve as biomarkers and warrant further research for potential drug target development.</p>\",\"PeriodicalId\":51650,\"journal\":{\"name\":\"BioMedicine-Taiwan\",\"volume\":\"14 4\",\"pages\":\"15-35\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11703398/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BioMedicine-Taiwan\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.37796/2211-8039.1467\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BioMedicine-Taiwan","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.37796/2211-8039.1467","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
摘要
背景:最具挑战性的癌症之一是三阴性乳腺癌,它被细分为许多分子亚型。由于高度的异质性,精准医疗的作用仍然具有挑战性。利用机器学习(ML)引导的基因选择,可以优化差异基因表达分析,最终通过生物标志物的发现,使精准医学的进程取得巨大进步。目的:通过鉴定最具代表性的差异表达基因作为生物标志物或新的药物靶点,加强肿瘤领域的精准医疗。方法:利用基因表达综合数据库(Gene Expression Omnibus, GEO)和癌症基因组图谱(the Cancer Genome Atlas, TCGA)的数据,利用线性微阵列分析模型(linear model for microarray analysis, LIMMA)和edgeR算法识别差异表达基因,并利用多种算法进行基于ml的特征选择。结果:通过融合基于LIMMA和基于ml的特征选择方法识别的特征,共选择了27个基因。曲线下面积(AUC)最高的模型是CatBoost、Extreme Gradient Boosting (XGBoost)、Random Forest和Multi-Layer Perceptron分类器。ESR1、FOXA1、GATA3、XBP1、GREB1、AR和AGR2在一个高度互联的集群中被鉴定为枢纽基因。结论:基于ml的基因选择对枢纽基因的鉴定有重要影响。所建立的机器学习模型可以提高肿瘤的精确诊断和预后。所鉴定的枢纽基因可以作为生物标志物,为潜在的药物靶点开发提供进一步的研究。
Machine learning-guided differential gene expression analysis identifies a highly-connected seven-gene cluster in triple-negative breast cancer.
Background: One of the most challenging cancers is triple-negative breast cancer, which is subdivided into many molecular subtypes. Due to the high degree of heterogeneity, the role of precision medicine remains challenging. With the use of machine learning (ML)-guided gene selection, the differential gene expression analysis can be optimized, and eventually, the process of precision medicine can see great advancement through biomarker discovery.
Purpose: Enhancing precision medicine in the oncology field by identification of the most representative differentially-expressed genes to be used as biomarkers or as novel drug targets.
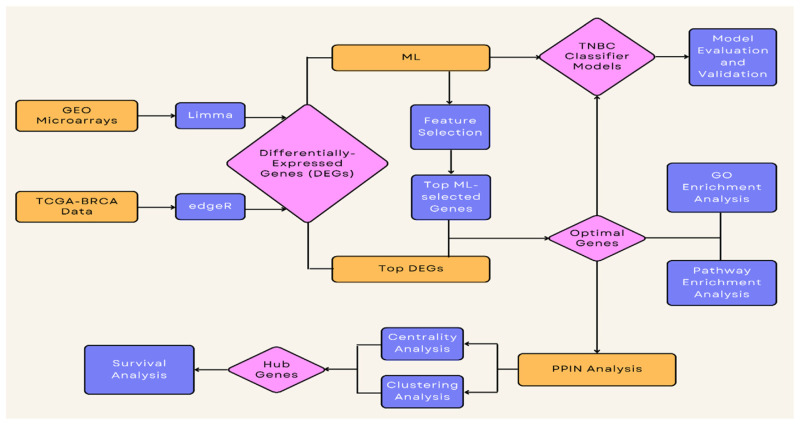
Methods: By utilizing data from the Gene Expression Omnibus (GEO) repository and The Cancer Genome Atlas (TCGA), we identified the differentially expressed genes using the linear model for microarray analysis (LIMMA) and edgeR algorithms, and applied ML-based feature selection using several algorithms.
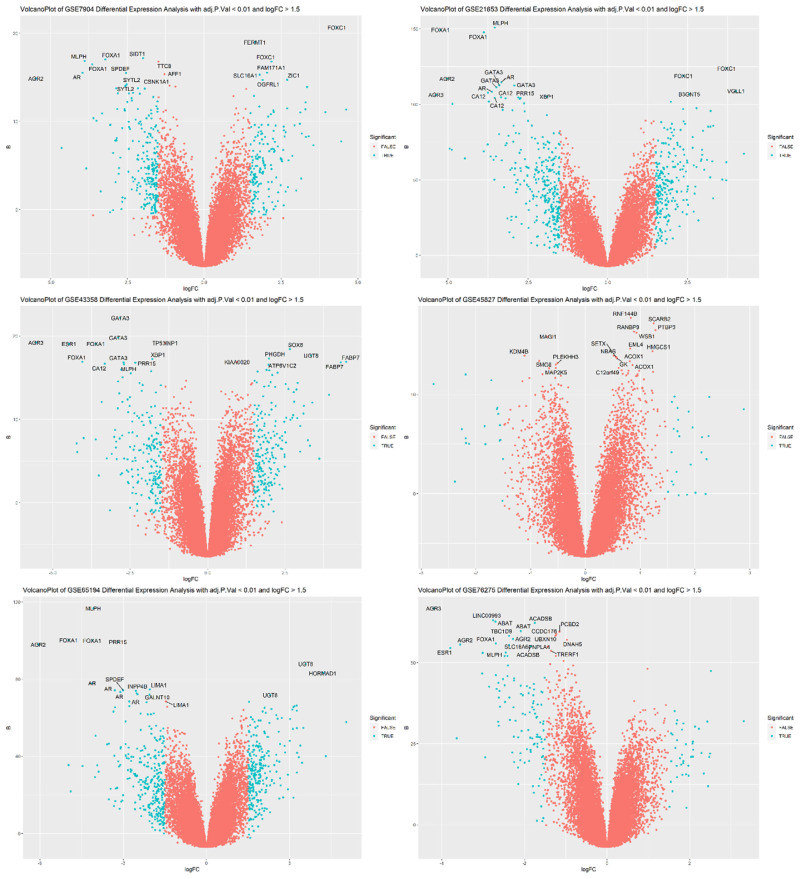
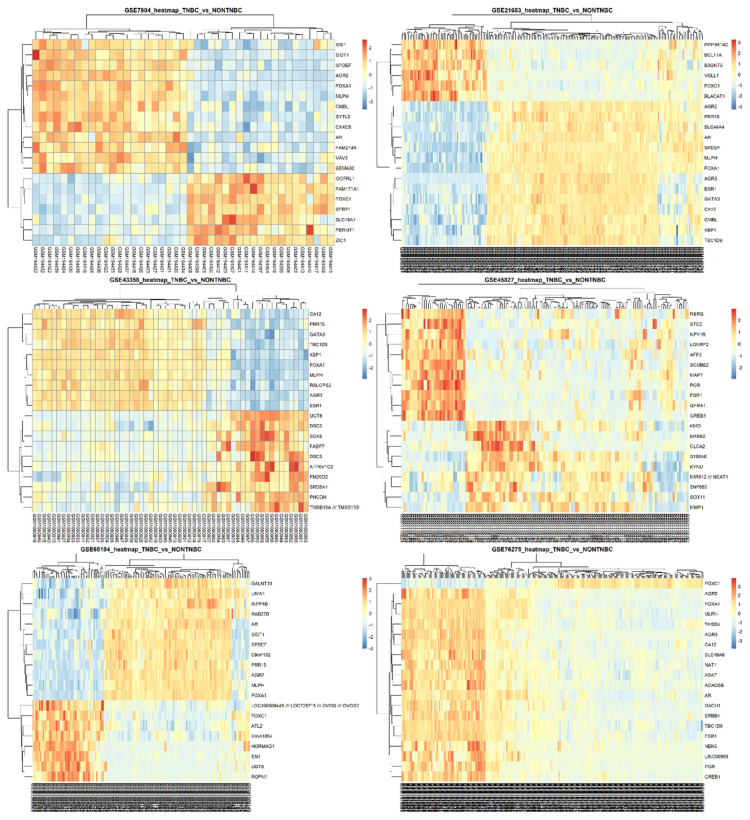
Results: A total of 27 genes were selected by merging features identified with both LIMMA and ML-based feature selection methods. The models with the highest area under the curve (AUC) are CatBoost, Extreme Gradient Boosting (XGBoost), Random Forest, and Multi-Layer Perceptron classifiers. ESR1, FOXA1, GATA3, XBP1, GREB1, AR, and AGR2 were identified as hub genes in a highly interconnected cluster.
Conclusion: ML-based gene selection shows a great impact on the identification of hub genes. The ML models built can improve precision oncology in diagnosis and prognosis. The identified hub genes can serve as biomarkers and warrant further research for potential drug target development.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


