Olusola Olagoke, Ammar Aziz, Lucile H Zhu, Timothy D Read, Deborah Dean
{"title":"利用集成的CtGAP管道对参考、体外和临床样品沙眼衣原体菌株进行全基因组自动化组装流水线。","authors":"Olusola Olagoke, Ammar Aziz, Lucile H Zhu, Timothy D Read, Deborah Dean","doi":"10.1093/nargab/lqae187","DOIUrl":null,"url":null,"abstract":"<p><p>Whole genome sequencing (WGS) is pivotal for the molecular characterization of <i>Chlamydia trachomatis</i> (<i>Ct</i>)-the leading bacterial cause of sexually transmitted infections and infectious blindness worldwide. <i>Ct</i> WGS can inform epidemiologic, public health and outbreak investigations of these human-restricted pathogens. However, challenges persist in generating high-quality genomes for downstream analyses given its obligate intracellular nature and difficulty with <i>in vitro</i> propagation. No single tool exists for the entirety of <i>Ct</i> genome assembly, necessitating the adaptation of multiple programs with varying success. Compounding this issue is the absence of reliable <i>Ct</i> reference strain genomes. We, therefore, developed CtGAP-<i>Chlamydia trachomatis</i>Genome Assembly Pipeline-as an integrated 'one-stop-shop' pipeline for assembly and characterization of <i>Ct</i> genome sequencing data from various sources including isolates, <i>in vitro</i> samples, clinical swabs and urine. CtGAP, written in Snakemake, enables read quality statistics output, adapter and quality trimming, host read removal, <i>de novo</i> and reference-guided assembly, contig scaffolding, selective <i>omp</i>A, multi-locus-sequence and plasmid typing, phylogenetic tree construction, and recombinant genome identification. Twenty <i>Ct</i> reference genomes were also generated. Successfully validated on a diverse collection of 363 samples containing <i>Ct</i>, CtGAP represents a novel pipeline requiring minimal bioinformatics expertise with easy adaptation for use with other bacterial species.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"7 1","pages":"lqae187"},"PeriodicalIF":2.8000,"publicationDate":"2025-01-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11704784/pdf/","citationCount":"0","resultStr":"{\"title\":\"Whole-genome automated assembly pipeline for <i>Chlamydia trachomatis</i> strains from reference, <i>in vitro</i> and clinical samples using the integrated CtGAP pipeline.\",\"authors\":\"Olusola Olagoke, Ammar Aziz, Lucile H Zhu, Timothy D Read, Deborah Dean\",\"doi\":\"10.1093/nargab/lqae187\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Whole genome sequencing (WGS) is pivotal for the molecular characterization of <i>Chlamydia trachomatis</i> (<i>Ct</i>)-the leading bacterial cause of sexually transmitted infections and infectious blindness worldwide. <i>Ct</i> WGS can inform epidemiologic, public health and outbreak investigations of these human-restricted pathogens. However, challenges persist in generating high-quality genomes for downstream analyses given its obligate intracellular nature and difficulty with <i>in vitro</i> propagation. No single tool exists for the entirety of <i>Ct</i> genome assembly, necessitating the adaptation of multiple programs with varying success. Compounding this issue is the absence of reliable <i>Ct</i> reference strain genomes. We, therefore, developed CtGAP-<i>Chlamydia trachomatis</i>Genome Assembly Pipeline-as an integrated 'one-stop-shop' pipeline for assembly and characterization of <i>Ct</i> genome sequencing data from various sources including isolates, <i>in vitro</i> samples, clinical swabs and urine. CtGAP, written in Snakemake, enables read quality statistics output, adapter and quality trimming, host read removal, <i>de novo</i> and reference-guided assembly, contig scaffolding, selective <i>omp</i>A, multi-locus-sequence and plasmid typing, phylogenetic tree construction, and recombinant genome identification. Twenty <i>Ct</i> reference genomes were also generated. Successfully validated on a diverse collection of 363 samples containing <i>Ct</i>, CtGAP represents a novel pipeline requiring minimal bioinformatics expertise with easy adaptation for use with other bacterial species.</p>\",\"PeriodicalId\":33994,\"journal\":{\"name\":\"NAR Genomics and Bioinformatics\",\"volume\":\"7 1\",\"pages\":\"lqae187\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-01-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11704784/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Genomics and Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/nargab/lqae187\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/3/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqae187","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Whole-genome automated assembly pipeline for Chlamydia trachomatis strains from reference, in vitro and clinical samples using the integrated CtGAP pipeline.
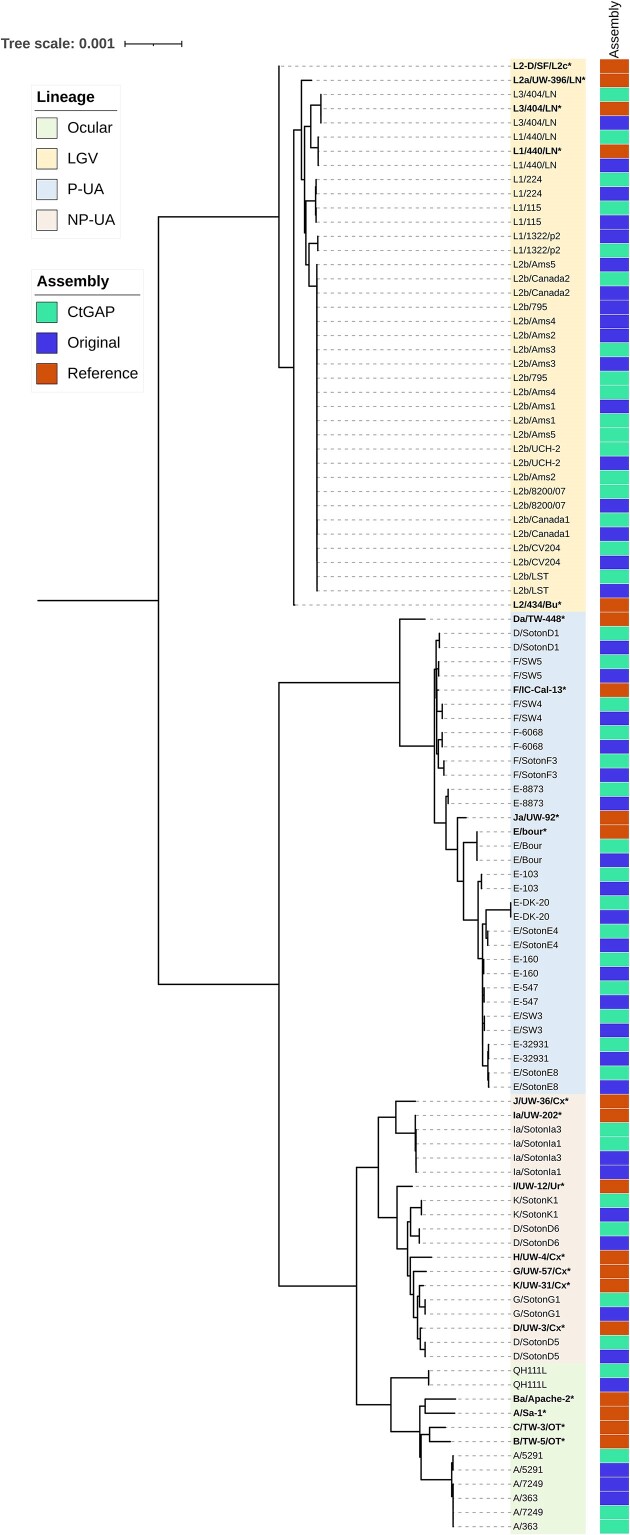
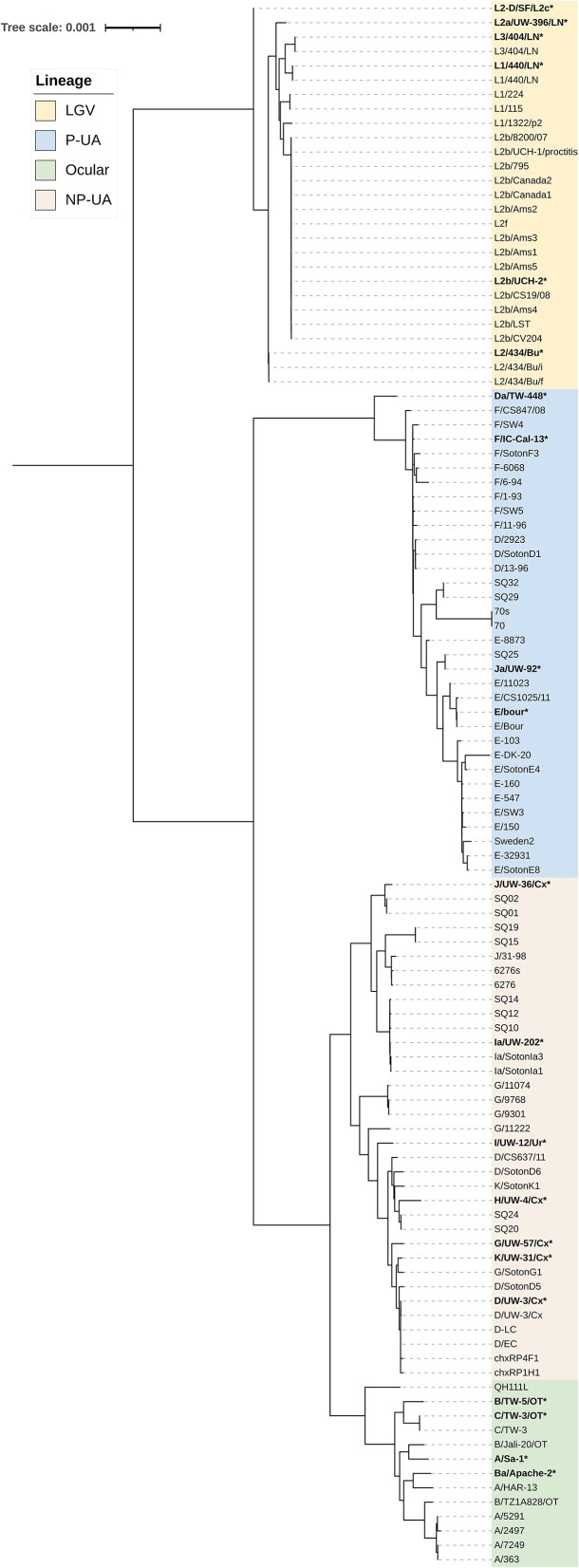
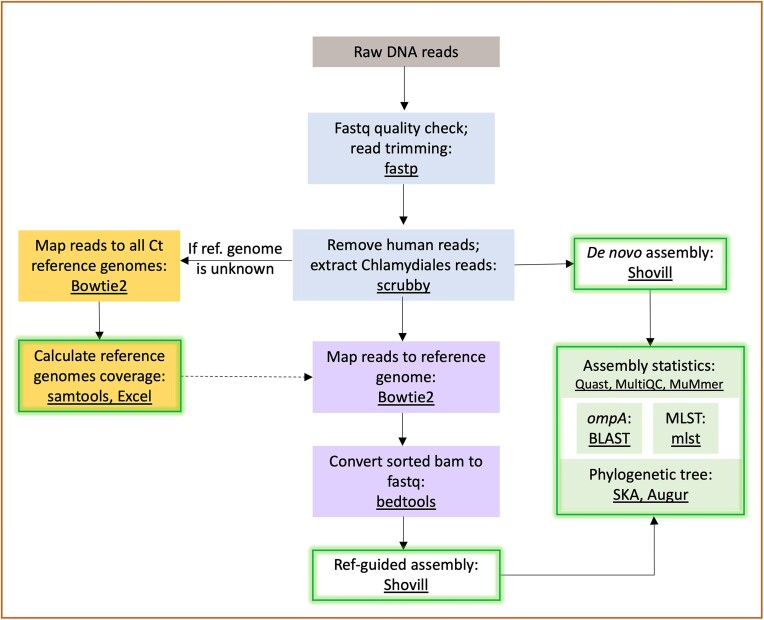
Whole genome sequencing (WGS) is pivotal for the molecular characterization of Chlamydia trachomatis (Ct)-the leading bacterial cause of sexually transmitted infections and infectious blindness worldwide. Ct WGS can inform epidemiologic, public health and outbreak investigations of these human-restricted pathogens. However, challenges persist in generating high-quality genomes for downstream analyses given its obligate intracellular nature and difficulty with in vitro propagation. No single tool exists for the entirety of Ct genome assembly, necessitating the adaptation of multiple programs with varying success. Compounding this issue is the absence of reliable Ct reference strain genomes. We, therefore, developed CtGAP-Chlamydia trachomatisGenome Assembly Pipeline-as an integrated 'one-stop-shop' pipeline for assembly and characterization of Ct genome sequencing data from various sources including isolates, in vitro samples, clinical swabs and urine. CtGAP, written in Snakemake, enables read quality statistics output, adapter and quality trimming, host read removal, de novo and reference-guided assembly, contig scaffolding, selective ompA, multi-locus-sequence and plasmid typing, phylogenetic tree construction, and recombinant genome identification. Twenty Ct reference genomes were also generated. Successfully validated on a diverse collection of 363 samples containing Ct, CtGAP represents a novel pipeline requiring minimal bioinformatics expertise with easy adaptation for use with other bacterial species.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


