{"title":"22q11.2微缺失引起的上颌中切牙孤立综合征。","authors":"Hirohito Shima, Akinobu Miura, Sayaka Kawashima, Ikumi Umeki, Chisumi Sogi, Dai Suzuki, Yusuke Takezawa, Ryo Sato, Natsuko Arai-Ichinoi, Miki Kamimura, Ikuma Fujiwara, Mika Adachi, Aya Yamada, Hiroshi Kawame, Atsuo Kikuchi, Junko Kanno","doi":"10.1297/cpe.2024-0024","DOIUrl":null,"url":null,"abstract":"<p><p>Solitary median maxillary central incisor (SMMCI) syndrome, the mildest form of the holoprosencephaly spectrum, is a rare anomaly characterized by the presence of a single midline central incisor in both the deciduous and permanent dentitions. Affected individuals can present with additional midline defects beyond dental findings. The 22q11.2 deletion syndrome (22q11.2 DS) arises from heterozygous microdeletions on chromosome 22q11.2, with breakpoints frequently located in eight clusters of low-copy repeats (LCR22A-H). Herein, we report an atypical case of 22q11.2 microdeletion in a male patient with SMMCI and additional features including hypothyroidism, ventricular septal defect, and several facial anomalies. The telomeric breakpoint was located in a segmental duplication 0.5 Mb distal to LCR22D, whereas the centromeric breakpoint was within LCR22C. Both segmental duplications shared a high level of sequence identity (97.2%), indicating the possibility of non-allelic homologous recombination (NAHR). This report supports the critical role of NAHR in the formation of rearrangements between regions other than LCR blocks and establishes a clinical association between 22q11.2 microdeletion and SMMCI.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"34 1","pages":"54-59"},"PeriodicalIF":1.2000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11701020/pdf/","citationCount":"0","resultStr":"{\"title\":\"Solitary median maxillary central incisor syndrome caused by 22q11.2 microdeletion.\",\"authors\":\"Hirohito Shima, Akinobu Miura, Sayaka Kawashima, Ikumi Umeki, Chisumi Sogi, Dai Suzuki, Yusuke Takezawa, Ryo Sato, Natsuko Arai-Ichinoi, Miki Kamimura, Ikuma Fujiwara, Mika Adachi, Aya Yamada, Hiroshi Kawame, Atsuo Kikuchi, Junko Kanno\",\"doi\":\"10.1297/cpe.2024-0024\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Solitary median maxillary central incisor (SMMCI) syndrome, the mildest form of the holoprosencephaly spectrum, is a rare anomaly characterized by the presence of a single midline central incisor in both the deciduous and permanent dentitions. Affected individuals can present with additional midline defects beyond dental findings. The 22q11.2 deletion syndrome (22q11.2 DS) arises from heterozygous microdeletions on chromosome 22q11.2, with breakpoints frequently located in eight clusters of low-copy repeats (LCR22A-H). Herein, we report an atypical case of 22q11.2 microdeletion in a male patient with SMMCI and additional features including hypothyroidism, ventricular septal defect, and several facial anomalies. The telomeric breakpoint was located in a segmental duplication 0.5 Mb distal to LCR22D, whereas the centromeric breakpoint was within LCR22C. Both segmental duplications shared a high level of sequence identity (97.2%), indicating the possibility of non-allelic homologous recombination (NAHR). This report supports the critical role of NAHR in the formation of rearrangements between regions other than LCR blocks and establishes a clinical association between 22q11.2 microdeletion and SMMCI.</p>\",\"PeriodicalId\":10678,\"journal\":{\"name\":\"Clinical Pediatric Endocrinology\",\"volume\":\"34 1\",\"pages\":\"54-59\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11701020/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1297/cpe.2024-0024\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2024-0024","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/12 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
孤立上颌正中切牙(SMMCI)综合征是一种罕见的异常,其特征是在乳牙和恒牙中都存在单个中线中切牙。受影响的个体可以呈现额外的中线缺陷超出牙科的发现。22q11.2缺失综合征(22q11.2 DS)是由染色体22q11.2的杂合微缺失引起的,断点通常位于8个低拷贝重复序列(lcr22 - a - h)。在此,我们报告了一例22q11.2微缺失的非典型病例,该患者患有SMMCI,其他特征包括甲状腺功能减退、室间隔缺损和一些面部异常。端粒断点位于LCR22D远端0.5 Mb的片段重复中,而着丝粒断点位于LCR22C内。两个片段重复具有较高的序列一致性(97.2%),表明存在非等位基因同源重组(NAHR)的可能性。该报告支持了NAHR在LCR区以外区域重排形成中的关键作用,并建立了22q11.2微缺失与SMMCI之间的临床关联。
Solitary median maxillary central incisor syndrome caused by 22q11.2 microdeletion.
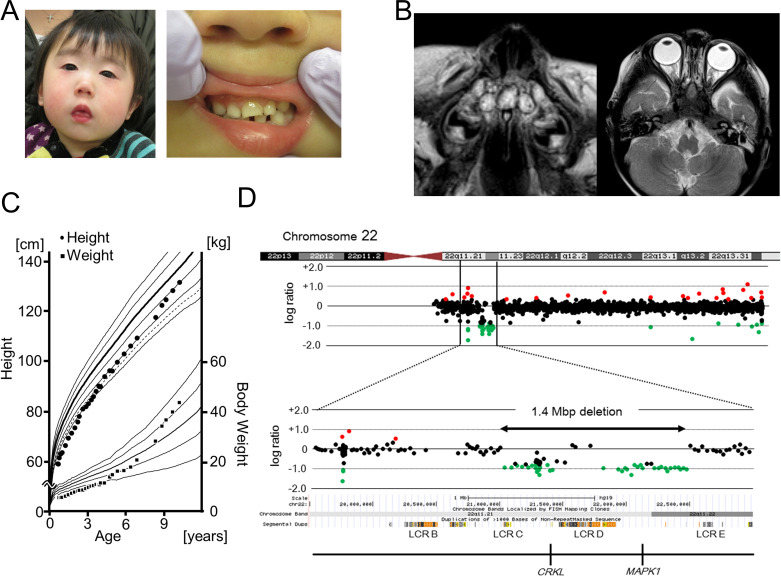
Solitary median maxillary central incisor (SMMCI) syndrome, the mildest form of the holoprosencephaly spectrum, is a rare anomaly characterized by the presence of a single midline central incisor in both the deciduous and permanent dentitions. Affected individuals can present with additional midline defects beyond dental findings. The 22q11.2 deletion syndrome (22q11.2 DS) arises from heterozygous microdeletions on chromosome 22q11.2, with breakpoints frequently located in eight clusters of low-copy repeats (LCR22A-H). Herein, we report an atypical case of 22q11.2 microdeletion in a male patient with SMMCI and additional features including hypothyroidism, ventricular septal defect, and several facial anomalies. The telomeric breakpoint was located in a segmental duplication 0.5 Mb distal to LCR22D, whereas the centromeric breakpoint was within LCR22C. Both segmental duplications shared a high level of sequence identity (97.2%), indicating the possibility of non-allelic homologous recombination (NAHR). This report supports the critical role of NAHR in the formation of rearrangements between regions other than LCR blocks and establishes a clinical association between 22q11.2 microdeletion and SMMCI.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


