{"title":"STLBRF:基于标准化阈值的改进型随机森林算法,用于基因表达数据的特征筛选。","authors":"Huini Feng, Ying Ju, Xiaofeng Yin, Wenshi Qiu, Xu Zhang","doi":"10.1093/bfgp/elae048","DOIUrl":null,"url":null,"abstract":"<p><p>When the traditional random forest (RF) algorithm is used to select feature elements in biostatistical data, a large amount of noise data and parameters can affect the importance of the selected feature elements, making the control of feature selection difficult. Therefore, it is a challenge for the traditional RF algorithm to preserve the accuracy of algorithm results in the presence of noise data. Generally, directly removing noise data can result in significant bias in the results. In this study, we develop a new algorithm, standardized threshold, and loops based random forest (STLBRF), and apply it to the field of gene expression data for feature gene selection. This algorithm, based on the traditional RF algorithm, combines backward elimination and K-fold cross-validation to construct a cyclic system and set a standardized threshold: error increment. The algorithm overcomes the shortcomings of existing gene selection methods. We compare ridge regression, lasso regression, elastic net regression, the traditional RF algorithm, and our improved RF algorithm using three real gene expression datasets and conducting a quantitative analysis. To ensure the reliability of the results, we validate the effectiveness of the genes selected by these methods using the Random Forest classifier. The results indicate that, compared to other methods, the STLBRF algorithm achieves not only higher effectiveness in feature gene selection but also better control over the number of selected genes. Our method offers reliable technical support for feature expression analysis and research on biomarker selection.</p>","PeriodicalId":55323,"journal":{"name":"Briefings in Functional Genomics","volume":" ","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11735748/pdf/","citationCount":"0","resultStr":"{\"title\":\"STLBRF: an improved random forest algorithm based on standardized-threshold for feature screening of gene expression data.\",\"authors\":\"Huini Feng, Ying Ju, Xiaofeng Yin, Wenshi Qiu, Xu Zhang\",\"doi\":\"10.1093/bfgp/elae048\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>When the traditional random forest (RF) algorithm is used to select feature elements in biostatistical data, a large amount of noise data and parameters can affect the importance of the selected feature elements, making the control of feature selection difficult. Therefore, it is a challenge for the traditional RF algorithm to preserve the accuracy of algorithm results in the presence of noise data. Generally, directly removing noise data can result in significant bias in the results. In this study, we develop a new algorithm, standardized threshold, and loops based random forest (STLBRF), and apply it to the field of gene expression data for feature gene selection. This algorithm, based on the traditional RF algorithm, combines backward elimination and K-fold cross-validation to construct a cyclic system and set a standardized threshold: error increment. The algorithm overcomes the shortcomings of existing gene selection methods. We compare ridge regression, lasso regression, elastic net regression, the traditional RF algorithm, and our improved RF algorithm using three real gene expression datasets and conducting a quantitative analysis. To ensure the reliability of the results, we validate the effectiveness of the genes selected by these methods using the Random Forest classifier. The results indicate that, compared to other methods, the STLBRF algorithm achieves not only higher effectiveness in feature gene selection but also better control over the number of selected genes. Our method offers reliable technical support for feature expression analysis and research on biomarker selection.</p>\",\"PeriodicalId\":55323,\"journal\":{\"name\":\"Briefings in Functional Genomics\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-01-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11735748/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Briefings in Functional Genomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bfgp/elae048\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in Functional Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bfgp/elae048","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
STLBRF: an improved random forest algorithm based on standardized-threshold for feature screening of gene expression data.
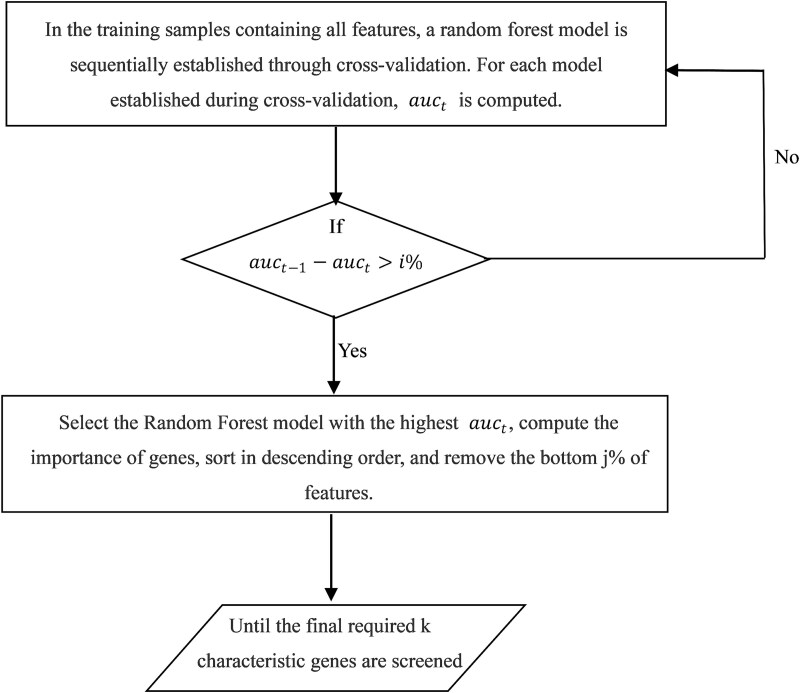
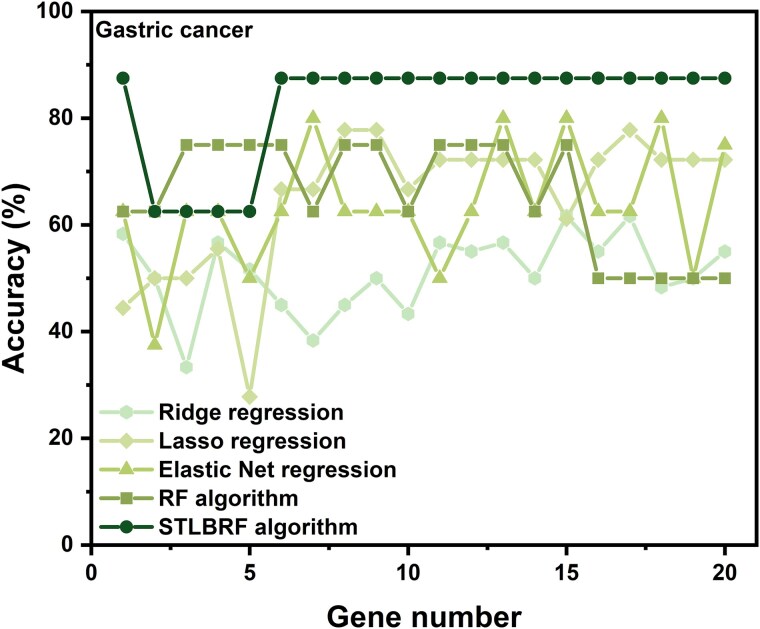
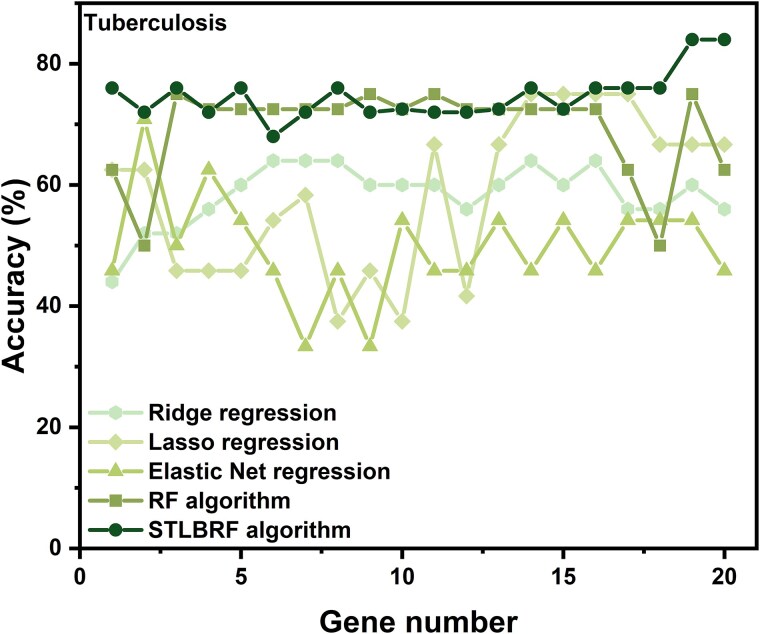
When the traditional random forest (RF) algorithm is used to select feature elements in biostatistical data, a large amount of noise data and parameters can affect the importance of the selected feature elements, making the control of feature selection difficult. Therefore, it is a challenge for the traditional RF algorithm to preserve the accuracy of algorithm results in the presence of noise data. Generally, directly removing noise data can result in significant bias in the results. In this study, we develop a new algorithm, standardized threshold, and loops based random forest (STLBRF), and apply it to the field of gene expression data for feature gene selection. This algorithm, based on the traditional RF algorithm, combines backward elimination and K-fold cross-validation to construct a cyclic system and set a standardized threshold: error increment. The algorithm overcomes the shortcomings of existing gene selection methods. We compare ridge regression, lasso regression, elastic net regression, the traditional RF algorithm, and our improved RF algorithm using three real gene expression datasets and conducting a quantitative analysis. To ensure the reliability of the results, we validate the effectiveness of the genes selected by these methods using the Random Forest classifier. The results indicate that, compared to other methods, the STLBRF algorithm achieves not only higher effectiveness in feature gene selection but also better control over the number of selected genes. Our method offers reliable technical support for feature expression analysis and research on biomarker selection.
期刊介绍:
Briefings in Functional Genomics publishes high quality peer reviewed articles that focus on the use, development or exploitation of genomic approaches, and their application to all areas of biological research. As well as exploring thematic areas where these techniques and protocols are being used, articles review the impact that these approaches have had, or are likely to have, on their field. Subjects covered by the Journal include but are not restricted to: the identification and functional characterisation of coding and non-coding features in genomes, microarray technologies, gene expression profiling, next generation sequencing, pharmacogenomics, phenomics, SNP technologies, transgenic systems, mutation screens and genotyping. Articles range in scope and depth from the introductory level to specific details of protocols and analyses, encompassing bacterial, fungal, plant, animal and human data.
The editorial board welcome the submission of review articles for publication. Essential criteria for the publication of papers is that they do not contain primary data, and that they are high quality, clearly written review articles which provide a balanced, highly informative and up to date perspective to researchers in the field of functional genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


