对沸石催化过程的现实模拟:一个小评论
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
沸石在工业催化中起着至关重要的作用,以其独特的微孔结构和多功能的催化性能而闻名。然而,由于其时空复杂性,准确模拟沸石催化过程面临着重大挑战,这需要捕获原子水平的相互作用和宏观现象。本文综述了沸石催化过程模拟的最新进展,重点介绍了机器学习电位(MLPs)、增强采样方法和动力学蒙特卡罗(KMC)模拟等技术。这些计算策略大大提高了催化反应模拟的准确性和效率,解决了沸石等复杂系统的传统局限性。mlp以较低的计算成本提供精确的势能面,实现扩展的分子动力学模拟。增强的采样技术,包括保护伞采样和元动力学,可以有效地探索罕见事件和复杂的能量景观,尽管它们的成功取决于对集体变量(cv)的仔细选择。KMC模拟通过在更大的空间和时间尺度上模拟长期的分子事件,如扩散和反应动力学,进一步增强了我们的理解。尽管取得了显著进展,但挑战依然存在,特别是在CV选择和KMC对精确第一原理数据的依赖方面。机器学习方法的集成,如自动CV选择和MLP优化的迁移学习,为这些问题提供了有希望的解决方案。这篇综述强调了这些进展及其对沸石催化过程的革命性研究的潜力,弥合了理论建模和实验观察之间的差距,并有助于设计更有效和可持续的催化剂。本文章由计算机程序翻译,如有差异,请以英文原文为准。
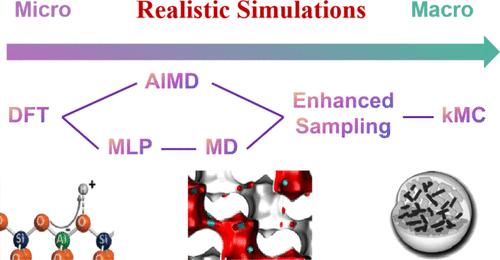
Toward Realistic Simulations of Zeolite Catalytic Processes: A Mini Review
Zeolites are crucial in industrial catalysis, renowned for their unique microporous structures and versatile catalytic properties. However, accurately simulating zeolite-catalyzed processes poses significant challenges due to their spatiotemporal complexity, which requires capturing both atomic-level interactions and macroscopic phenomena. This review examines recent advancements in realistic simulations of zeolite catalytic processes, focusing on techniques such as machine learning potentials (MLPs), enhanced sampling methods, and kinetic Monte Carlo (KMC) simulations. These computational strategies have substantially improved the accuracy and efficiency of catalytic reaction simulations, addressing the traditional limitations associated with complex systems like zeolites. MLPs offer precise potential energy surfaces at lower computational costs, enabling extended molecular dynamics simulations. Enhanced sampling techniques, including umbrella sampling and metadynamics, effectively explore rare events and complex energy landscapes, although their success depends on the careful selection of collective variables (CVs). KMC simulations further enhance our understanding by modeling long-term molecular events, such as diffusion and reaction kinetics, at larger spatial and temporal scales. Despite notable progress, challenges remain, particularly regarding CV selection and KMC’s reliance on accurate first-principles data. The integration of machine learning approaches, such as automated CV selection and transfer learning for MLP refinement, presents promising solutions to these issues. This review highlights these advancements and their potential to revolutionize the study of zeolite catalytic processes, bridging the gap between theoretical modeling and experimental observations and contributing to the design of more effective and sustainable catalysts.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


