Federica Barzaghi, Mattia Moratti, Giuseppina Panza, Beatrice Rivalta, Giuliana Giardino, Antonio De Rosa, Lucia Augusta Baselli, Matteo Chinello, Antonio Marzollo, Davide Montin, Maddalena Marinoni, Giorgio Costagliola, Silvia Ricci, Lorenzo Lodi, Baldassarre Martire, Cinzia Milito, Antonino Trizzino, Alberto Tommasini, Marco Zecca, Raffaele Badolato, Caterina Cancrini, Vassilios Lougaris, Claudio Pignata, Francesca Conti
{"title":"靶向治疗时代活化磷酸肌肽3-激酶δ综合征的意大利队列报告。","authors":"Federica Barzaghi, Mattia Moratti, Giuseppina Panza, Beatrice Rivalta, Giuliana Giardino, Antonio De Rosa, Lucia Augusta Baselli, Matteo Chinello, Antonio Marzollo, Davide Montin, Maddalena Marinoni, Giorgio Costagliola, Silvia Ricci, Lorenzo Lodi, Baldassarre Martire, Cinzia Milito, Antonino Trizzino, Alberto Tommasini, Marco Zecca, Raffaele Badolato, Caterina Cancrini, Vassilios Lougaris, Claudio Pignata, Francesca Conti","doi":"10.1007/s10875-024-01835-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Activated Phosphoinositide 3-Kinase (PI3K) δ Syndrome (APDS), an inborn error of immunity due to upregulation of the PI3K pathway, leads to recurrent infections and immune dysregulation (lymphoproliferation and autoimmunity).</p><p><strong>Methods: </strong>Clinical and genetic data of 28 APDS patients from 25 unrelated families were collected from fifteen Italian centers.</p><p><strong>Results: </strong>Patients were genetically confirmed with APDS-1 (n = 20) or APDS-2 (n = 8), with pathogenic mutations in the PIK3CD or PIK3R1 genes. The median age at diagnosis was 15.5 years, with a median follow-up of 74 months (range 6-384). The main presenting symptoms were respiratory tract infections alone (57%) or associated with lymphoproliferation (17%). Later, non-clonal lymphoproliferation was the leading clinical sign (86%), followed by respiratory infections (79%) and gastrointestinal complications (43%). Malignant lymphoproliferative disorders, all EBV-encoding RNA (EBER)-positive at the histological analysis, occurred in 14% of patients aged 17-19 years, highlighting the role of EBV in lymphomagenesis in this disorder. Diffuse large B-cell lymphoma was the most frequent. Immunological work-up revealed combined T/B cell abnormalities in most patients. Treatment strategies included immunosuppression and PI3K/Akt/mTOR inhibitor therapy. Rapamycin, employed in 36% of patients, showed efficacy in controlling lymphoproliferation, while selective PI3Kδ inhibitor leniolisib, administered in 32% of patients, was beneficial on both infections and immune dysregulation. Additionally, three patients underwent successful HSCT due to recurrent infections despite ongoing prophylaxis or lymphoproliferation poorly responsive to Rapamycin.</p><p><strong>Conclusions: </strong>This study underscores the clinical heterogeneity and challenging diagnosis of APDS, highlighting the importance of multidisciplinary management tailored to individual needs and further supporting leniolisib efficacy.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"58"},"PeriodicalIF":5.7000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11666751/pdf/","citationCount":"0","resultStr":"{\"title\":\"Report of the Italian Cohort with Activated Phosphoinositide 3-Kinase δ Syndrome in the Target Therapy Era.\",\"authors\":\"Federica Barzaghi, Mattia Moratti, Giuseppina Panza, Beatrice Rivalta, Giuliana Giardino, Antonio De Rosa, Lucia Augusta Baselli, Matteo Chinello, Antonio Marzollo, Davide Montin, Maddalena Marinoni, Giorgio Costagliola, Silvia Ricci, Lorenzo Lodi, Baldassarre Martire, Cinzia Milito, Antonino Trizzino, Alberto Tommasini, Marco Zecca, Raffaele Badolato, Caterina Cancrini, Vassilios Lougaris, Claudio Pignata, Francesca Conti\",\"doi\":\"10.1007/s10875-024-01835-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Activated Phosphoinositide 3-Kinase (PI3K) δ Syndrome (APDS), an inborn error of immunity due to upregulation of the PI3K pathway, leads to recurrent infections and immune dysregulation (lymphoproliferation and autoimmunity).</p><p><strong>Methods: </strong>Clinical and genetic data of 28 APDS patients from 25 unrelated families were collected from fifteen Italian centers.</p><p><strong>Results: </strong>Patients were genetically confirmed with APDS-1 (n = 20) or APDS-2 (n = 8), with pathogenic mutations in the PIK3CD or PIK3R1 genes. The median age at diagnosis was 15.5 years, with a median follow-up of 74 months (range 6-384). The main presenting symptoms were respiratory tract infections alone (57%) or associated with lymphoproliferation (17%). Later, non-clonal lymphoproliferation was the leading clinical sign (86%), followed by respiratory infections (79%) and gastrointestinal complications (43%). Malignant lymphoproliferative disorders, all EBV-encoding RNA (EBER)-positive at the histological analysis, occurred in 14% of patients aged 17-19 years, highlighting the role of EBV in lymphomagenesis in this disorder. Diffuse large B-cell lymphoma was the most frequent. Immunological work-up revealed combined T/B cell abnormalities in most patients. Treatment strategies included immunosuppression and PI3K/Akt/mTOR inhibitor therapy. Rapamycin, employed in 36% of patients, showed efficacy in controlling lymphoproliferation, while selective PI3Kδ inhibitor leniolisib, administered in 32% of patients, was beneficial on both infections and immune dysregulation. Additionally, three patients underwent successful HSCT due to recurrent infections despite ongoing prophylaxis or lymphoproliferation poorly responsive to Rapamycin.</p><p><strong>Conclusions: </strong>This study underscores the clinical heterogeneity and challenging diagnosis of APDS, highlighting the importance of multidisciplinary management tailored to individual needs and further supporting leniolisib efficacy.</p>\",\"PeriodicalId\":15531,\"journal\":{\"name\":\"Journal of Clinical Immunology\",\"volume\":\"45 1\",\"pages\":\"58\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-12-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11666751/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10875-024-01835-1\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-024-01835-1","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Report of the Italian Cohort with Activated Phosphoinositide 3-Kinase δ Syndrome in the Target Therapy Era.
Background: Activated Phosphoinositide 3-Kinase (PI3K) δ Syndrome (APDS), an inborn error of immunity due to upregulation of the PI3K pathway, leads to recurrent infections and immune dysregulation (lymphoproliferation and autoimmunity).
Methods: Clinical and genetic data of 28 APDS patients from 25 unrelated families were collected from fifteen Italian centers.
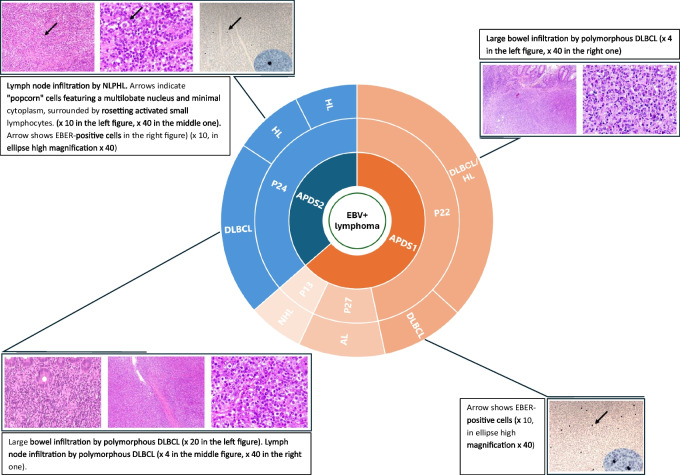
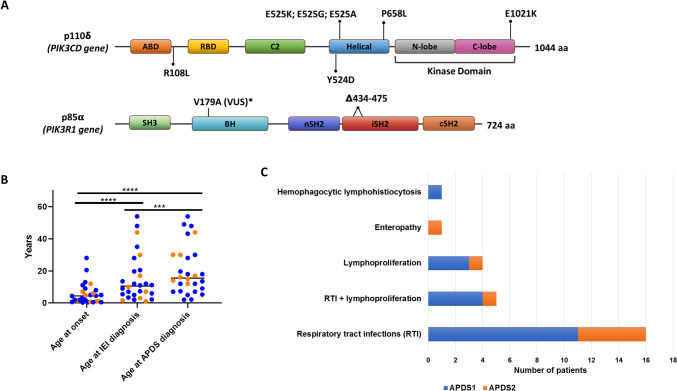
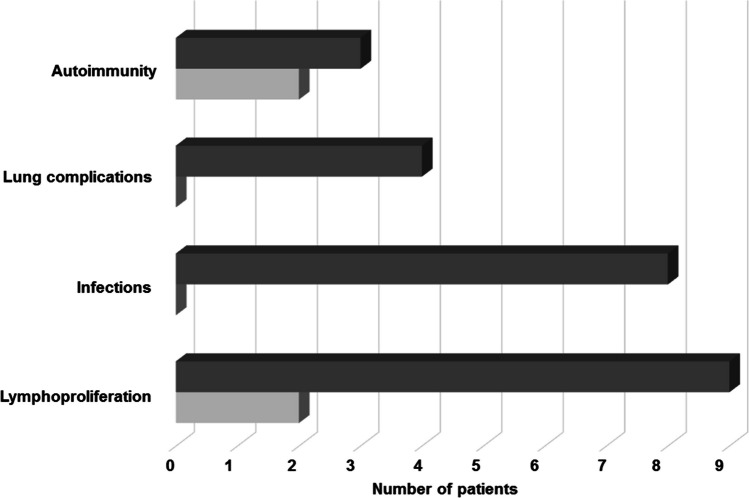
Results: Patients were genetically confirmed with APDS-1 (n = 20) or APDS-2 (n = 8), with pathogenic mutations in the PIK3CD or PIK3R1 genes. The median age at diagnosis was 15.5 years, with a median follow-up of 74 months (range 6-384). The main presenting symptoms were respiratory tract infections alone (57%) or associated with lymphoproliferation (17%). Later, non-clonal lymphoproliferation was the leading clinical sign (86%), followed by respiratory infections (79%) and gastrointestinal complications (43%). Malignant lymphoproliferative disorders, all EBV-encoding RNA (EBER)-positive at the histological analysis, occurred in 14% of patients aged 17-19 years, highlighting the role of EBV in lymphomagenesis in this disorder. Diffuse large B-cell lymphoma was the most frequent. Immunological work-up revealed combined T/B cell abnormalities in most patients. Treatment strategies included immunosuppression and PI3K/Akt/mTOR inhibitor therapy. Rapamycin, employed in 36% of patients, showed efficacy in controlling lymphoproliferation, while selective PI3Kδ inhibitor leniolisib, administered in 32% of patients, was beneficial on both infections and immune dysregulation. Additionally, three patients underwent successful HSCT due to recurrent infections despite ongoing prophylaxis or lymphoproliferation poorly responsive to Rapamycin.
Conclusions: This study underscores the clinical heterogeneity and challenging diagnosis of APDS, highlighting the importance of multidisciplinary management tailored to individual needs and further supporting leniolisib efficacy.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


