Thomas Cassini, Sarah Silverstein, Molly Behan, Cynthia J. Tifft, May Christine Malicdan, David R. Adams, Undiagnosed Diseases Network, Sun-Young Ahn, Debra S. Regier
{"title":"深层内含子缺失导致的线粒体三功能蛋白缺陷导致异常剪接","authors":"Thomas Cassini, Sarah Silverstein, Molly Behan, Cynthia J. Tifft, May Christine Malicdan, David R. Adams, Undiagnosed Diseases Network, Sun-Young Ahn, Debra S. Regier","doi":"10.1002/jmd2.12459","DOIUrl":null,"url":null,"abstract":"<p>Trifunctional protein deficiency (TFP) is a disorder of fatty acid beta-oxidation associated with metabolic, cardiac, and liver dysfunction in severe forms. We present two siblings diagnosed by newborn screening and confirmed by biochemical testing at birth. Their clinical course was complicated by recurrent rhabdomyolysis, retinopathy, and hypoparathyroidism. Both siblings were also diagnosed with focal segmental glomerulosclerosis (FSGS) and bone marrow failure and ultimately died of hypoxemic respiratory failure. Initial sequencing of the TFP-associated genes <i>HADHA</i> and <i>HADHB</i> showed only a paternally inherited variant in <i>HADHB,</i> NM_000183.3:c.1059del (p.Gly354AspfsTer10). Subsequent evaluation by the Undiagnosed Diseases Network with genome and transcriptome sequencing revealed a rare maternally inherited 17 base pair deletion in <i>HADHB</i>, NM_000183.3:c.1390-515_1390-499del, located in the final intron and resulting in a pseudoexon that harbors a premature termination codon. Both sisters were compound heterozygous for this and the paternal premature termination codon. No other variants were detected that were potentially causative for the FSGS and bone marrow failure on genome sequencing. A review of the literature at that time revealed several case reports of the uncommon clinical findings of FSGS, bone marrow failure, and pulmonary involvement in patients with TFP, confirming this clinical diagnosis as the complete explanation for these siblings.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"66 1","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-12-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12459","citationCount":"0","resultStr":"{\"title\":\"Mitochondrial trifunctional protein deficiency caused by a deep intronic deletion leading to aberrant splicing\",\"authors\":\"Thomas Cassini, Sarah Silverstein, Molly Behan, Cynthia J. Tifft, May Christine Malicdan, David R. Adams, Undiagnosed Diseases Network, Sun-Young Ahn, Debra S. Regier\",\"doi\":\"10.1002/jmd2.12459\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Trifunctional protein deficiency (TFP) is a disorder of fatty acid beta-oxidation associated with metabolic, cardiac, and liver dysfunction in severe forms. We present two siblings diagnosed by newborn screening and confirmed by biochemical testing at birth. Their clinical course was complicated by recurrent rhabdomyolysis, retinopathy, and hypoparathyroidism. Both siblings were also diagnosed with focal segmental glomerulosclerosis (FSGS) and bone marrow failure and ultimately died of hypoxemic respiratory failure. Initial sequencing of the TFP-associated genes <i>HADHA</i> and <i>HADHB</i> showed only a paternally inherited variant in <i>HADHB,</i> NM_000183.3:c.1059del (p.Gly354AspfsTer10). Subsequent evaluation by the Undiagnosed Diseases Network with genome and transcriptome sequencing revealed a rare maternally inherited 17 base pair deletion in <i>HADHB</i>, NM_000183.3:c.1390-515_1390-499del, located in the final intron and resulting in a pseudoexon that harbors a premature termination codon. Both sisters were compound heterozygous for this and the paternal premature termination codon. No other variants were detected that were potentially causative for the FSGS and bone marrow failure on genome sequencing. A review of the literature at that time revealed several case reports of the uncommon clinical findings of FSGS, bone marrow failure, and pulmonary involvement in patients with TFP, confirming this clinical diagnosis as the complete explanation for these siblings.</p>\",\"PeriodicalId\":14930,\"journal\":{\"name\":\"JIMD reports\",\"volume\":\"66 1\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-12-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12459\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JIMD reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12459\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12459","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Mitochondrial trifunctional protein deficiency caused by a deep intronic deletion leading to aberrant splicing
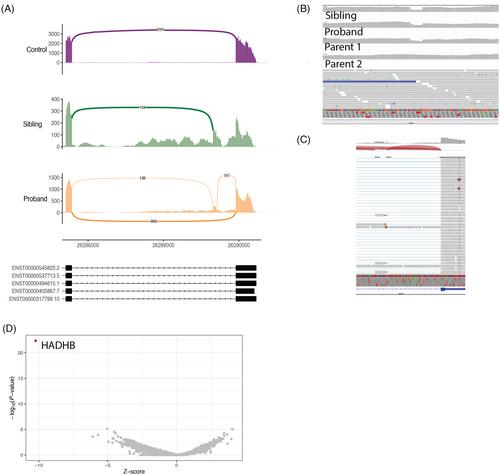
Trifunctional protein deficiency (TFP) is a disorder of fatty acid beta-oxidation associated with metabolic, cardiac, and liver dysfunction in severe forms. We present two siblings diagnosed by newborn screening and confirmed by biochemical testing at birth. Their clinical course was complicated by recurrent rhabdomyolysis, retinopathy, and hypoparathyroidism. Both siblings were also diagnosed with focal segmental glomerulosclerosis (FSGS) and bone marrow failure and ultimately died of hypoxemic respiratory failure. Initial sequencing of the TFP-associated genes HADHA and HADHB showed only a paternally inherited variant in HADHB, NM_000183.3:c.1059del (p.Gly354AspfsTer10). Subsequent evaluation by the Undiagnosed Diseases Network with genome and transcriptome sequencing revealed a rare maternally inherited 17 base pair deletion in HADHB, NM_000183.3:c.1390-515_1390-499del, located in the final intron and resulting in a pseudoexon that harbors a premature termination codon. Both sisters were compound heterozygous for this and the paternal premature termination codon. No other variants were detected that were potentially causative for the FSGS and bone marrow failure on genome sequencing. A review of the literature at that time revealed several case reports of the uncommon clinical findings of FSGS, bone marrow failure, and pulmonary involvement in patients with TFP, confirming this clinical diagnosis as the complete explanation for these siblings.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


