Ahmed Sarar Mohamed, Talal AlAnzi, Amal Alhashem, Hadeel Alrukban, Fahad Al Harbi, Sarar Mohamed
{"title":"沙特阿拉伯典型同型胱氨酸尿症的临床、生化和分子特征,以及新生儿筛查对预防并发症的影响:一家三级医疗中心的经验","authors":"Ahmed Sarar Mohamed, Talal AlAnzi, Amal Alhashem, Hadeel Alrukban, Fahad Al Harbi, Sarar Mohamed","doi":"10.1002/jmd2.12454","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Classic homocystinuria (HCU) is a rare inborn metabolic disease that is generally asymptomatic at birth. If untreated, it can cause a wide range of complications including intellectual disability, lens dislocation, and thromboembolism. This study aimed to describe the natural history and the molecular findings of patients with HCU, and to assess the importance of early diagnosis.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>This study retrospectively collected data on patients attending the metabolic unit at Prince Sultan Military Medical City, Riyadh, Saudi Arabia from 2011 to 2024. Demographic, clinical, and molecular data was extracted from the electronic medical records.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Among the 33 patients with HCU enrolled, 5/33 (15%) were diagnosed by newborn screening and the rest were diagnosed on clinical grounds. The complication profile was vast, with neuropsychiatric, musculoskeletal, ophthalmic, and thromboembolic morbidities complicating the disease course in 28/33 (85%) of the patients. None of the newborn screened patients had complications while all of the non-newborn screened patients had at least one complication, <i>p</i> < 0.0001. The majority of parents in this cohort were highly consanguineous, with 90% had first or second cousin marriage. Seven previously reported variants were detected in this cohort and one novel variant was found in three patients (c.828+2-828+ 3 delins ACACTTGCATCC, p.?). The known pathogenic variant (c.969G>A, p. (Trp323*)) was seen in most of the patients, with all of them coming from one tribe.</p>\n </section>\n \n <section>\n \n <h3> Conclusions</h3>\n \n <p>This cohort gives further evidence that the newborn screening for HCU is likely to prevent the complications associated with the disease at least in the first few years of life. Therefore, newborn screening for HCU should be encouraged. Our molecular studies revealed the presence of a founder variant, detected in patients from a single tribe. This suggests that specific mutation testing may be cost-effective for individuals from certain ethnicities.</p>\n </section>\n </div>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"66 1","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-11-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12454","citationCount":"0","resultStr":"{\"title\":\"Clinical, biochemical and molecular characteristics of classic homocystinuria in Saudi Arabia and the impact of newborn screening on prevention of the complications: A tertiary center experience\",\"authors\":\"Ahmed Sarar Mohamed, Talal AlAnzi, Amal Alhashem, Hadeel Alrukban, Fahad Al Harbi, Sarar Mohamed\",\"doi\":\"10.1002/jmd2.12454\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Classic homocystinuria (HCU) is a rare inborn metabolic disease that is generally asymptomatic at birth. If untreated, it can cause a wide range of complications including intellectual disability, lens dislocation, and thromboembolism. This study aimed to describe the natural history and the molecular findings of patients with HCU, and to assess the importance of early diagnosis.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>This study retrospectively collected data on patients attending the metabolic unit at Prince Sultan Military Medical City, Riyadh, Saudi Arabia from 2011 to 2024. Demographic, clinical, and molecular data was extracted from the electronic medical records.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Among the 33 patients with HCU enrolled, 5/33 (15%) were diagnosed by newborn screening and the rest were diagnosed on clinical grounds. The complication profile was vast, with neuropsychiatric, musculoskeletal, ophthalmic, and thromboembolic morbidities complicating the disease course in 28/33 (85%) of the patients. None of the newborn screened patients had complications while all of the non-newborn screened patients had at least one complication, <i>p</i> < 0.0001. The majority of parents in this cohort were highly consanguineous, with 90% had first or second cousin marriage. Seven previously reported variants were detected in this cohort and one novel variant was found in three patients (c.828+2-828+ 3 delins ACACTTGCATCC, p.?). The known pathogenic variant (c.969G>A, p. (Trp323*)) was seen in most of the patients, with all of them coming from one tribe.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusions</h3>\\n \\n <p>This cohort gives further evidence that the newborn screening for HCU is likely to prevent the complications associated with the disease at least in the first few years of life. Therefore, newborn screening for HCU should be encouraged. Our molecular studies revealed the presence of a founder variant, detected in patients from a single tribe. This suggests that specific mutation testing may be cost-effective for individuals from certain ethnicities.</p>\\n </section>\\n </div>\",\"PeriodicalId\":14930,\"journal\":{\"name\":\"JIMD reports\",\"volume\":\"66 1\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-11-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12454\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JIMD reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12454\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12454","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Clinical, biochemical and molecular characteristics of classic homocystinuria in Saudi Arabia and the impact of newborn screening on prevention of the complications: A tertiary center experience
Background
Classic homocystinuria (HCU) is a rare inborn metabolic disease that is generally asymptomatic at birth. If untreated, it can cause a wide range of complications including intellectual disability, lens dislocation, and thromboembolism. This study aimed to describe the natural history and the molecular findings of patients with HCU, and to assess the importance of early diagnosis.
Methods
This study retrospectively collected data on patients attending the metabolic unit at Prince Sultan Military Medical City, Riyadh, Saudi Arabia from 2011 to 2024. Demographic, clinical, and molecular data was extracted from the electronic medical records.
Results
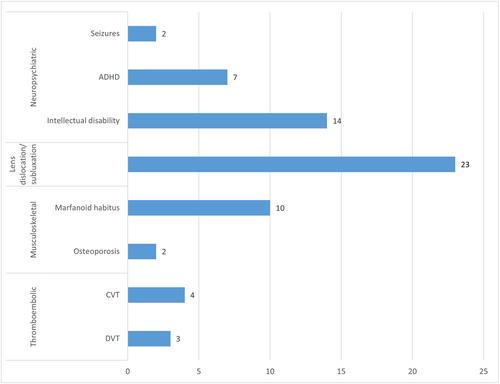
Among the 33 patients with HCU enrolled, 5/33 (15%) were diagnosed by newborn screening and the rest were diagnosed on clinical grounds. The complication profile was vast, with neuropsychiatric, musculoskeletal, ophthalmic, and thromboembolic morbidities complicating the disease course in 28/33 (85%) of the patients. None of the newborn screened patients had complications while all of the non-newborn screened patients had at least one complication, p < 0.0001. The majority of parents in this cohort were highly consanguineous, with 90% had first or second cousin marriage. Seven previously reported variants were detected in this cohort and one novel variant was found in three patients (c.828+2-828+ 3 delins ACACTTGCATCC, p.?). The known pathogenic variant (c.969G>A, p. (Trp323*)) was seen in most of the patients, with all of them coming from one tribe.
Conclusions
This cohort gives further evidence that the newborn screening for HCU is likely to prevent the complications associated with the disease at least in the first few years of life. Therefore, newborn screening for HCU should be encouraged. Our molecular studies revealed the presence of a founder variant, detected in patients from a single tribe. This suggests that specific mutation testing may be cost-effective for individuals from certain ethnicities.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


