Jakub Horvath, Pavel Jedlicka, Marie Kratka, Zdenek Kubat, Eduard Kejnovsky, Matej Lexa
{"title":"植物ltr -反转录转座子长末端重复序列的检测和分类及其可解释性机器学习分析。","authors":"Jakub Horvath, Pavel Jedlicka, Marie Kratka, Zdenek Kubat, Eduard Kejnovsky, Matej Lexa","doi":"10.1186/s13040-024-00410-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Long terminal repeats (LTRs) represent important parts of LTR retrotransposons and retroviruses found in high copy numbers in a majority of eukaryotic genomes. LTRs contain regulatory sequences essential for the life cycle of the retrotransposon. Previous experimental and sequence studies have provided only limited information about LTR structure and composition, mostly from model systems. To enhance our understanding of these key sequence modules, we focused on the contrasts between LTRs of various retrotransposon families and other genomic regions. Furthermore, this approach can be utilized for the classification and prediction of LTRs.</p><p><strong>Results: </strong>We used machine learning methods suitable for DNA sequence classification and applied them to a large dataset of plant LTR retrotransposon sequences. We trained three machine learning models using (i) traditional model ensembles (Gradient Boosting), (ii) hybrid convolutional/long and short memory network models, and (iii) a DNA pre-trained transformer-based model using k-mer sequence representation. All three approaches were successful in classifying and isolating LTRs in this data, as well as providing valuable insights into LTR sequence composition. The best classification (expressed as F1 score) achieved for LTR detection was 0.85 using the hybrid network model. The most accurate classification task was superfamily classification (F1=0.89) while the least accurate was family classification (F1=0.74). The trained models were subjected to explainability analysis. Positional analysis identified a mixture of interesting features, many of which had a preferred absolute position within the LTR and/or were biologically relevant, such as a centrally positioned TATA-box regulatory sequence, and TG..CA nucleotide patterns around both LTR edges.</p><p><strong>Conclusions: </strong>Our results show that the models used here recognized biologically relevant motifs, such as core promoter elements in the LTR detection task, and a development and stress-related subclass of transcription factor binding sites in the family classification task. Explainability analysis also highlighted the importance of 5'- and 3'- edges in LTR identity and revealed need to analyze more than just dinucleotides at these ends. Our work shows the applicability of machine learning models to regulatory sequence analysis and classification, and demonstrates the important role of the identified motifs in LTR detection.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"17 1","pages":"57"},"PeriodicalIF":6.1000,"publicationDate":"2024-12-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11656987/pdf/","citationCount":"0","resultStr":"{\"title\":\"Detection and classification of long terminal repeat sequences in plant LTR-retrotransposons and their analysis using explainable machine learning.\",\"authors\":\"Jakub Horvath, Pavel Jedlicka, Marie Kratka, Zdenek Kubat, Eduard Kejnovsky, Matej Lexa\",\"doi\":\"10.1186/s13040-024-00410-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Long terminal repeats (LTRs) represent important parts of LTR retrotransposons and retroviruses found in high copy numbers in a majority of eukaryotic genomes. LTRs contain regulatory sequences essential for the life cycle of the retrotransposon. Previous experimental and sequence studies have provided only limited information about LTR structure and composition, mostly from model systems. To enhance our understanding of these key sequence modules, we focused on the contrasts between LTRs of various retrotransposon families and other genomic regions. Furthermore, this approach can be utilized for the classification and prediction of LTRs.</p><p><strong>Results: </strong>We used machine learning methods suitable for DNA sequence classification and applied them to a large dataset of plant LTR retrotransposon sequences. We trained three machine learning models using (i) traditional model ensembles (Gradient Boosting), (ii) hybrid convolutional/long and short memory network models, and (iii) a DNA pre-trained transformer-based model using k-mer sequence representation. All three approaches were successful in classifying and isolating LTRs in this data, as well as providing valuable insights into LTR sequence composition. The best classification (expressed as F1 score) achieved for LTR detection was 0.85 using the hybrid network model. The most accurate classification task was superfamily classification (F1=0.89) while the least accurate was family classification (F1=0.74). The trained models were subjected to explainability analysis. Positional analysis identified a mixture of interesting features, many of which had a preferred absolute position within the LTR and/or were biologically relevant, such as a centrally positioned TATA-box regulatory sequence, and TG..CA nucleotide patterns around both LTR edges.</p><p><strong>Conclusions: </strong>Our results show that the models used here recognized biologically relevant motifs, such as core promoter elements in the LTR detection task, and a development and stress-related subclass of transcription factor binding sites in the family classification task. Explainability analysis also highlighted the importance of 5'- and 3'- edges in LTR identity and revealed need to analyze more than just dinucleotides at these ends. Our work shows the applicability of machine learning models to regulatory sequence analysis and classification, and demonstrates the important role of the identified motifs in LTR detection.</p>\",\"PeriodicalId\":48947,\"journal\":{\"name\":\"Biodata Mining\",\"volume\":\"17 1\",\"pages\":\"57\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2024-12-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11656987/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biodata Mining\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13040-024-00410-z\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-024-00410-z","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Detection and classification of long terminal repeat sequences in plant LTR-retrotransposons and their analysis using explainable machine learning.
Background: Long terminal repeats (LTRs) represent important parts of LTR retrotransposons and retroviruses found in high copy numbers in a majority of eukaryotic genomes. LTRs contain regulatory sequences essential for the life cycle of the retrotransposon. Previous experimental and sequence studies have provided only limited information about LTR structure and composition, mostly from model systems. To enhance our understanding of these key sequence modules, we focused on the contrasts between LTRs of various retrotransposon families and other genomic regions. Furthermore, this approach can be utilized for the classification and prediction of LTRs.
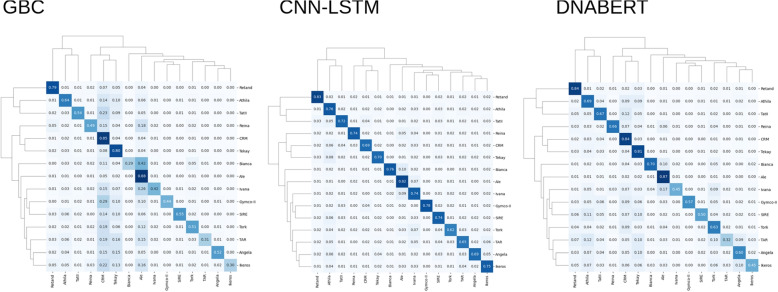
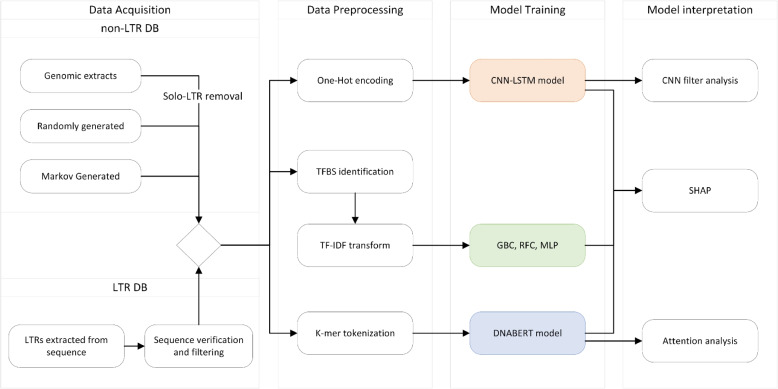
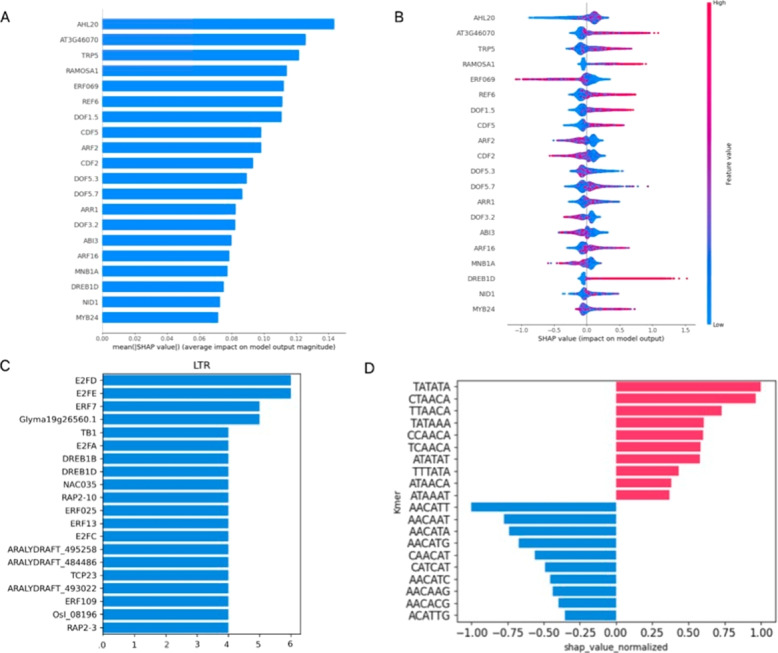
Results: We used machine learning methods suitable for DNA sequence classification and applied them to a large dataset of plant LTR retrotransposon sequences. We trained three machine learning models using (i) traditional model ensembles (Gradient Boosting), (ii) hybrid convolutional/long and short memory network models, and (iii) a DNA pre-trained transformer-based model using k-mer sequence representation. All three approaches were successful in classifying and isolating LTRs in this data, as well as providing valuable insights into LTR sequence composition. The best classification (expressed as F1 score) achieved for LTR detection was 0.85 using the hybrid network model. The most accurate classification task was superfamily classification (F1=0.89) while the least accurate was family classification (F1=0.74). The trained models were subjected to explainability analysis. Positional analysis identified a mixture of interesting features, many of which had a preferred absolute position within the LTR and/or were biologically relevant, such as a centrally positioned TATA-box regulatory sequence, and TG..CA nucleotide patterns around both LTR edges.
Conclusions: Our results show that the models used here recognized biologically relevant motifs, such as core promoter elements in the LTR detection task, and a development and stress-related subclass of transcription factor binding sites in the family classification task. Explainability analysis also highlighted the importance of 5'- and 3'- edges in LTR identity and revealed need to analyze more than just dinucleotides at these ends. Our work shows the applicability of machine learning models to regulatory sequence analysis and classification, and demonstrates the important role of the identified motifs in LTR detection.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


