IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
二氧化硅(SiO2)表面原子氧(O)催化重组系数的测量值和计算值存在相当大的不确定性,这对准确预测加热负荷,进而对大气高超声速飞行器进行重量效益设计构成了巨大挑战。这项研究基于 ReaxFFSiOGSI 进行了大规模(以反应轨迹计)反应分子动力学模拟,ReaxFF 是专为 O(气体)-SiO2(表面)相互作用定制的 ReaxFF 势函数,旨在了解不同 SiO2 表面结构下 O 重组的化学过程。通过 O 在相同 SiO2 表面结构上的吸附进行密度函数理论计算,验证了本基于 ReaxFF 的分子动力学的适用性。为了从大量的轨迹中捕捉反应路径和机理,我们开发了一种自动数据分析器。研究发现,吸附、活性位点形成和重组的途径对表面结构非常敏感。在不同的表面结构下,不同反应途径的总体重组系数及其组成差异很大。我们首次发现了一种涉及多个活性位点的反应机制,这种机制比单位点反应更容易发生,因此有可能增加重组概率。这些发现凸显了表面结构在催化重组反应中的重要作用,并为以往研究中重组系数的巨大差异提供了可能的解释。本文章由计算机程序翻译,如有差异,请以英文原文为准。
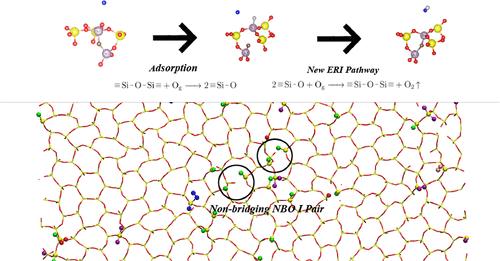
Uncover Chemical Processes for Silica Surfaces Exposed to Atomic Oxygen Using ReaxFF Reactive Molecular Dynamics
The considerable level of uncertainty in the measured and calculated catalytic recombination coefficients of atomic oxygen (O) on silica (SiO2) surfaces has posed a great challenge to the accurate prediction of heating load and thereby the weight-effective design for atmospheric hypersonic vehicles. This work conducts large-scale (in terms of reaction trajectories) reactive molecular dynamics simulations based on ReaxFFSiOGSI, a ReaxFF potential function tailored for O(gas)-SiO2(surface) interactions to understand the chemical processes for the recombination of O for different SiO2 surface structures. The applicability of the present ReaxFF-based molecular dynamics is validated by the density-functional-theory calculation through O adsorption on the same SiO2 surface structures under investigation. An automatic data analyzer is developed to capture the reaction pathways and mechanisms from the vast amount of trajectories. It is found that the pathways of adsorption, active site formation, and recombination are sensitive to the surface structures. The overall recombination coefficient and its compositions from different reaction pathways vary considerably for different surface structures. We identify for the first time a reaction mechanism involving multiple active sites, which is more likely to occur than the single-site reactions and thus can potentially increase the recombination probability. These findings highlight the important role of surface structure in catalytic recombination reactions and provide a possible explanation for the huge discrepancy in the recombination coefficients from previous studies.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


