Frederieke Lohmann, Stephan Allenspach, Kenneth Atz, Carl C G Schiebroek, Jan A Hiss, Gisbert Schneider
{"title":"潜空间中的蛋白质结合位点表征","authors":"Frederieke Lohmann, Stephan Allenspach, Kenneth Atz, Carl C G Schiebroek, Jan A Hiss, Gisbert Schneider","doi":"10.1002/minf.202400205","DOIUrl":null,"url":null,"abstract":"<p><p>Interpretability and reliability of deep learning models are important for computer-based drug discovery. Aiming to understand feature perception by such a model, we investigate a graph neural network for affinity prediction of protein-ligand complexes. We assess a latent representation of ligand binding sites and investigate underlying geometric structure in this latent space and its relation to protein function. We introduce an automated computational pipeline for dimensionality reduction, clustering, hypothesis testing, and visualization of latent space. The results indicate that the learned protein latent space is inherently structured and not randomly distributed. Several of the identified protein binding site clusters in latent space correspond to functional protein families. Ligand size was found to be a determinant of cluster geometry. The computational pipeline proved applicable to latent space analysis and interpretation and can be adapted to work for different datasets and deep learning models.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":" ","pages":"e202400205"},"PeriodicalIF":3.1000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11733832/pdf/","citationCount":"0","resultStr":"{\"title\":\"Protein Binding Site Representation in Latent Space.\",\"authors\":\"Frederieke Lohmann, Stephan Allenspach, Kenneth Atz, Carl C G Schiebroek, Jan A Hiss, Gisbert Schneider\",\"doi\":\"10.1002/minf.202400205\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Interpretability and reliability of deep learning models are important for computer-based drug discovery. Aiming to understand feature perception by such a model, we investigate a graph neural network for affinity prediction of protein-ligand complexes. We assess a latent representation of ligand binding sites and investigate underlying geometric structure in this latent space and its relation to protein function. We introduce an automated computational pipeline for dimensionality reduction, clustering, hypothesis testing, and visualization of latent space. The results indicate that the learned protein latent space is inherently structured and not randomly distributed. Several of the identified protein binding site clusters in latent space correspond to functional protein families. Ligand size was found to be a determinant of cluster geometry. The computational pipeline proved applicable to latent space analysis and interpretation and can be adapted to work for different datasets and deep learning models.</p>\",\"PeriodicalId\":18853,\"journal\":{\"name\":\"Molecular Informatics\",\"volume\":\" \",\"pages\":\"e202400205\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11733832/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Informatics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/minf.202400205\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/12/18 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202400205","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/18 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Protein Binding Site Representation in Latent Space.
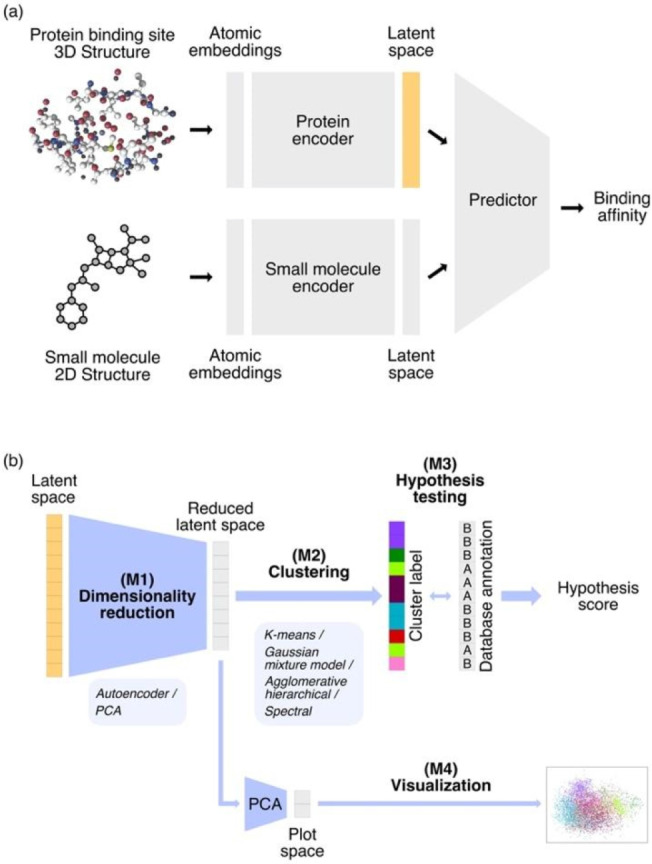
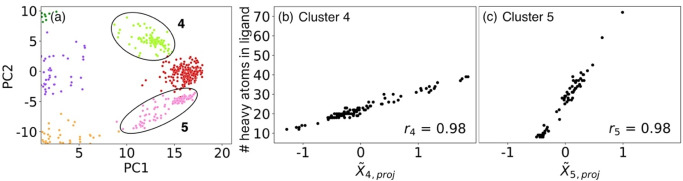
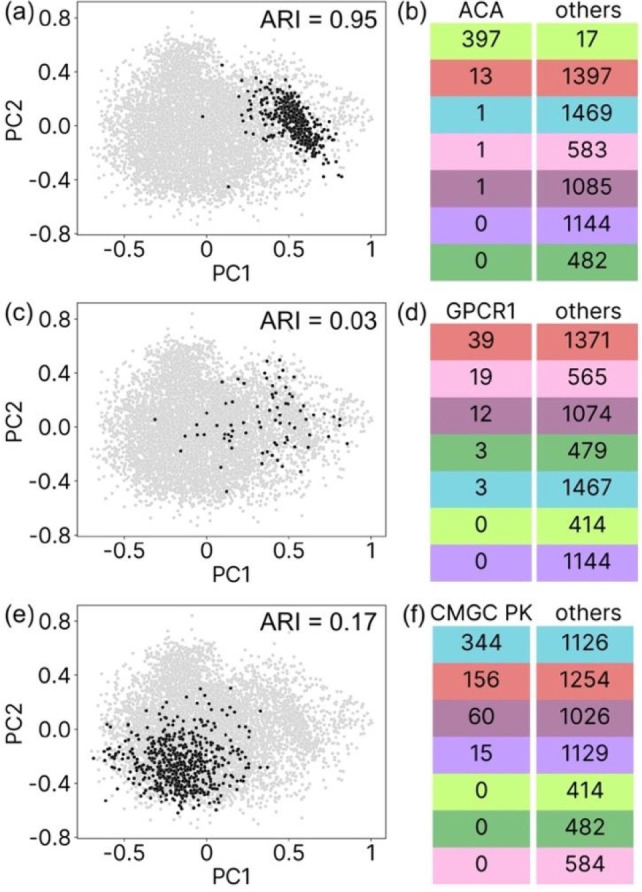
Interpretability and reliability of deep learning models are important for computer-based drug discovery. Aiming to understand feature perception by such a model, we investigate a graph neural network for affinity prediction of protein-ligand complexes. We assess a latent representation of ligand binding sites and investigate underlying geometric structure in this latent space and its relation to protein function. We introduce an automated computational pipeline for dimensionality reduction, clustering, hypothesis testing, and visualization of latent space. The results indicate that the learned protein latent space is inherently structured and not randomly distributed. Several of the identified protein binding site clusters in latent space correspond to functional protein families. Ligand size was found to be a determinant of cluster geometry. The computational pipeline proved applicable to latent space analysis and interpretation and can be adapted to work for different datasets and deep learning models.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


