高保真机器学习原子间势能的数据高效多保真度训练
IF 15.6
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
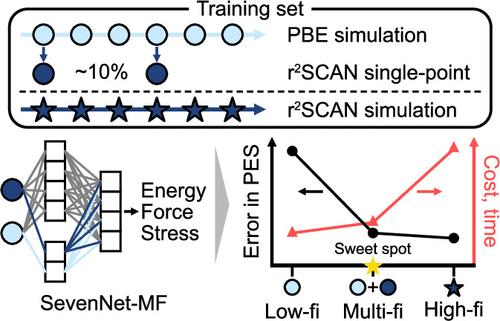
机器学习原子间势能(MLIPs)用于估算来自原子序数计算的势能面(PES),在降低计算成本的同时提供接近量子级的精确度。然而,组建高保真数据库的高成本阻碍了 MLIPs 在需要高化学精度的系统中的应用。利用等变图神经网络,我们提出了一种可同时在多保真度数据库上进行训练的 MLIP 框架。这种方法能以最少的高保真数据准确学习高保真 PES。我们分别采用广义梯度逼近(GGA)和元 GGA 作为低保真和高保真方法,在 Li6PS5Cl 和 InxGa1-xN 系统上测试了这一框架。结果表明,使用大小约为低保真集 10% 的高保真训练集,多保真训练框架实现了极高的准确性,与参考的高保真 MLIP 结果相比,锂离子电导率预测误差在 10% 以内,InxGa1-xN 混合能的 R2 值为 0.98。这表明,高保真元 GGA 数据库未覆盖的几何和成分空间可以有效地从低保真 GGA 数据中推断出来,从而提高准确性和分子动力学稳定性。我们还开发了一种通用 MLIP,可同时利用材料项目中的 GGA 和元 GGA 数据,大大提高了 MLIP 在高精度任务(如预测一般晶体的赫尔以上能量)中的性能。此外,我们还证明了目前的多保真度学习比迁移学习或Δ学习更有效,而且还可以应用于耦合簇水平的更高保真度学习。我们相信,这种方法有望通过有效扩展高保真数据集,创建高精度的定制或通用 MLIP。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Data-Efficient Multifidelity Training for High-Fidelity Machine Learning Interatomic Potentials
Machine learning interatomic potentials (MLIPs) are used to estimate potential energy surfaces (PES) from ab initio calculations, providing near-quantum-level accuracy with reduced computational costs. However, the high cost of assembling high-fidelity databases hampers the application of MLIPs to systems that require high chemical accuracy. Utilizing an equivariant graph neural network, we present an MLIP framework that trains on multifidelity databases simultaneously. This approach enables the accurate learning of high-fidelity PES with minimal high-fidelity data. Employing the generalized gradient approximation (GGA) and meta-GGA as low- and high-fidelity approaches, respectively, we tested this framework on the Li6PS5Cl and InxGa1–xN systems. The results show that using a high-fidelity training set with a size approximately 10% of the low-fidelity set, the multifidelity training framework achieves excellent accuracy, with Li-ion conductivity predictions within 10% error and InxGa1–xN mixing energy showing an R2 of 0.98 compared to the reference high-fidelity MLIP results. It indicates that geometric and compositional spaces not covered by the high-fidelity meta-GGA database can be effectively inferred from low-fidelity GGA data, thus enhancing accuracy and molecular dynamics stability. We also developed a general-purpose MLIP that utilizes both GGA and meta-GGA data from the Materials Project, significantly enhancing MLIP performance for high-accuracy tasks such as predicting energies above hull for crystals in general. Furthermore, we demonstrate that the present multifidelity learning is more effective than transfer learning or Δ-learning and that it can also be applied to learn higher-fidelity up to the coupled-cluster level. We believe this methodology holds promise for creating highly accurate bespoke or universal MLIPs by effectively expanding the high-fidelity data set.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


