Laura Alfonso-Hernandez, Victor M. Freixas, Tammie Gibson, Sergei Tretiak, Sebastian Fernandez-Alberti
{"title":"通过纳米环的联锁调谐纳米环的电子松弛","authors":"Laura Alfonso-Hernandez, Victor M. Freixas, Tammie Gibson, Sergei Tretiak, Sebastian Fernandez-Alberti","doi":"10.1002/jcc.27533","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Electronic and vibrational relaxation processes can be optimized and tuned by introducing alternative pathways that channel excess energy more efficiently. An ensemble of interacting molecular systems can help overcome the bottlenecks caused by large energy gaps between intermediate excited states involved in the relaxation process. By employing this strategy, catenanes composed of mechanically interlocked carbon nanostructures show great promise as new materials for achieving higher efficiencies in electronic devices. Herein, we perform nonadiabatic excited state molecular dynamics on different all-benzene catenanes. We observe that catenanes experience faster relaxations than individual units. Coupled catenanes present overlapping energy manifolds that include several electronic excited states spatially localized on the different moieties, increasing the density of states that ultimately improve the efficiency in the energy relaxation. This result suggests the use of catenanes as a viable strategy for tuning the internal conversion rates in a quest for their utilization for new optoelectronic applications.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Tuning Electronic Relaxation of Nanorings Through Their Interlocking\",\"authors\":\"Laura Alfonso-Hernandez, Victor M. Freixas, Tammie Gibson, Sergei Tretiak, Sebastian Fernandez-Alberti\",\"doi\":\"10.1002/jcc.27533\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>Electronic and vibrational relaxation processes can be optimized and tuned by introducing alternative pathways that channel excess energy more efficiently. An ensemble of interacting molecular systems can help overcome the bottlenecks caused by large energy gaps between intermediate excited states involved in the relaxation process. By employing this strategy, catenanes composed of mechanically interlocked carbon nanostructures show great promise as new materials for achieving higher efficiencies in electronic devices. Herein, we perform nonadiabatic excited state molecular dynamics on different all-benzene catenanes. We observe that catenanes experience faster relaxations than individual units. Coupled catenanes present overlapping energy manifolds that include several electronic excited states spatially localized on the different moieties, increasing the density of states that ultimately improve the efficiency in the energy relaxation. This result suggests the use of catenanes as a viable strategy for tuning the internal conversion rates in a quest for their utilization for new optoelectronic applications.</p>\\n </div>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-12-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27533\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27533","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Tuning Electronic Relaxation of Nanorings Through Their Interlocking
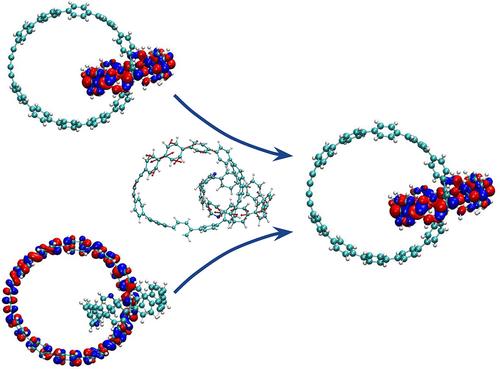
Electronic and vibrational relaxation processes can be optimized and tuned by introducing alternative pathways that channel excess energy more efficiently. An ensemble of interacting molecular systems can help overcome the bottlenecks caused by large energy gaps between intermediate excited states involved in the relaxation process. By employing this strategy, catenanes composed of mechanically interlocked carbon nanostructures show great promise as new materials for achieving higher efficiencies in electronic devices. Herein, we perform nonadiabatic excited state molecular dynamics on different all-benzene catenanes. We observe that catenanes experience faster relaxations than individual units. Coupled catenanes present overlapping energy manifolds that include several electronic excited states spatially localized on the different moieties, increasing the density of states that ultimately improve the efficiency in the energy relaxation. This result suggests the use of catenanes as a viable strategy for tuning the internal conversion rates in a quest for their utilization for new optoelectronic applications.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


