NiCoFeAlTiB多主元素合金的有序/无序相变:蒙特卡罗分析
IF 9.3
1区 材料科学
Q1 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
多主元素固溶体容易产生局部化学不均匀性,例如,化学有序/聚类和/或成分波动。为了模拟复杂多主元合金(mpea)的结构转变,建立了一种柔性双能量模型。晶格蒙特卡罗(MC)模拟,结合离散原子对能量,广泛地研究了nicofe基合金的热力学原子结构。模拟结果与报道的实验数据一致,并提供了相分解,物种分离/偏好和化学有序/无序的见解。模拟还通过在晶界处加入小原子(如硼),提供了原子偏析的微观视图。采用卡恩润湿理论,对观察到的相变过程,从完全润湿到部分/预润湿,进行了热力学解释和验证。目前的方法有可能扩展到各种具有恒定晶格的MPEA系统。本文章由计算机程序翻译,如有差异,请以英文原文为准。

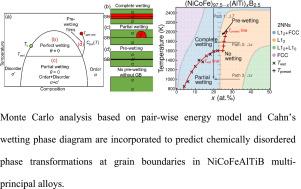
Chemical order/disorder phase transitions in NiCoFeAlTiB multi-principal element alloys: A Monte Carlo analysis
Multi-principal element solid solutions are prone to develop local chemical inhomogeneities, e.g., chemical order/clustering and/or compositional undulation. To model the structural transformation of complex multi-principal element alloys (MPEAs), a flexible pairwise-energy model is developed. Lattice Monte Carlo (MC) simulations, coupled with discrete atomic pair energies, are extensively conducted to investigate thermodynamic atomic structures of NiCoFe-based alloys. The results of simulations are consistent with reported experimental data and provide insights into phase decomposition, species segregation/preferences, and chemical order/disorder. The simulations also offer microscopic views of atomic segregation by incorporating small atom (e.g., Boron) at grain boundaries. By employing Cahn’s wetting theory, the observed phase transformation processes, ranging from perfect to partial/pre-wetting, are explained and validated thermodynamically by the model. The current approach has the potential to be extended to a variety of MPEA systems with constant lattices.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Acta Materialia
工程技术-材料科学:综合
CiteScore
16.10
自引率
8.50%
发文量
801
审稿时长
53 days
期刊介绍:
Acta Materialia serves as a platform for publishing full-length, original papers and commissioned overviews that contribute to a profound understanding of the correlation between the processing, structure, and properties of inorganic materials. The journal seeks papers with high impact potential or those that significantly propel the field forward. The scope includes the atomic and molecular arrangements, chemical and electronic structures, and microstructure of materials, focusing on their mechanical or functional behavior across all length scales, including nanostructures.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


