Dipanshu Kumar, Joachim Sauer, Alessia Airi, Silvia Bordiga and Daria Ruth Galimberti
{"title":"用费米共振模型确定H-ZSM-5 Brønsted位点乙醇的红外光谱与单体吸附的关系","authors":"Dipanshu Kumar, Joachim Sauer, Alessia Airi, Silvia Bordiga and Daria Ruth Galimberti","doi":"10.1039/D4CP03861D","DOIUrl":null,"url":null,"abstract":"<p >Understanding how alcohol molecules interact with the Brønsted acid sites (BAS) of zeolites is a prerequisite to the design of zeolite catalysts and catalytic processes. Here, we report IR spectra for the adsorption of ethanol on a highly crystalline sample of H-ZSM-5 zeolites exposed to ethanol gas at increasing pressure. We use density functional theory in combination with a FERMI resonance model to assign the measured spectra to a single adsorbed ethanol molecule per BAS. Specifically, we assign the bands at 2450 cm<small><sup>−1</sup></small> and 1670 cm<small><sup>−1</sup></small> to a FERMI resonance between the fundamental (Z)O–H stretching band of a single-ethanol-loaded BAS and the first overtone of the (Z)O–H out-of-plane bending. We conclude that adsorbed dimers do not contribute in a noticeable way up to a concentration of almost one ethanol molecule per BAS site. We further show that hybrid functionals (B3LYP) are required to get a close match between the predicted and experimental spectra, whereas commonly used generalized gradient type functionals such as PBE incorrectly describe the potential energy surface. They overestimate the redshift of the OH stretching band on hydrogen bond formation which results in an erroneous assignment of the IR bands.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 1","pages":" 550-563"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp03861d?page=search","citationCount":"0","resultStr":"{\"title\":\"Assignment of IR spectra of ethanol at Brønsted sites of H-ZSM-5 to monomer adsorption using a Fermi resonance model†\",\"authors\":\"Dipanshu Kumar, Joachim Sauer, Alessia Airi, Silvia Bordiga and Daria Ruth Galimberti\",\"doi\":\"10.1039/D4CP03861D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Understanding how alcohol molecules interact with the Brønsted acid sites (BAS) of zeolites is a prerequisite to the design of zeolite catalysts and catalytic processes. Here, we report IR spectra for the adsorption of ethanol on a highly crystalline sample of H-ZSM-5 zeolites exposed to ethanol gas at increasing pressure. We use density functional theory in combination with a FERMI resonance model to assign the measured spectra to a single adsorbed ethanol molecule per BAS. Specifically, we assign the bands at 2450 cm<small><sup>−1</sup></small> and 1670 cm<small><sup>−1</sup></small> to a FERMI resonance between the fundamental (Z)O–H stretching band of a single-ethanol-loaded BAS and the first overtone of the (Z)O–H out-of-plane bending. We conclude that adsorbed dimers do not contribute in a noticeable way up to a concentration of almost one ethanol molecule per BAS site. We further show that hybrid functionals (B3LYP) are required to get a close match between the predicted and experimental spectra, whereas commonly used generalized gradient type functionals such as PBE incorrectly describe the potential energy surface. They overestimate the redshift of the OH stretching band on hydrogen bond formation which results in an erroneous assignment of the IR bands.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 1\",\"pages\":\" 550-563\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-12-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp03861d?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03861d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03861d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Assignment of IR spectra of ethanol at Brønsted sites of H-ZSM-5 to monomer adsorption using a Fermi resonance model†
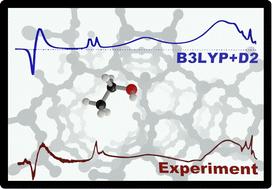
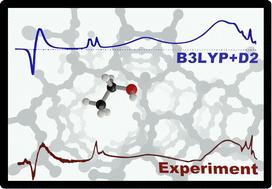
Understanding how alcohol molecules interact with the Brønsted acid sites (BAS) of zeolites is a prerequisite to the design of zeolite catalysts and catalytic processes. Here, we report IR spectra for the adsorption of ethanol on a highly crystalline sample of H-ZSM-5 zeolites exposed to ethanol gas at increasing pressure. We use density functional theory in combination with a FERMI resonance model to assign the measured spectra to a single adsorbed ethanol molecule per BAS. Specifically, we assign the bands at 2450 cm−1 and 1670 cm−1 to a FERMI resonance between the fundamental (Z)O–H stretching band of a single-ethanol-loaded BAS and the first overtone of the (Z)O–H out-of-plane bending. We conclude that adsorbed dimers do not contribute in a noticeable way up to a concentration of almost one ethanol molecule per BAS site. We further show that hybrid functionals (B3LYP) are required to get a close match between the predicted and experimental spectra, whereas commonly used generalized gradient type functionals such as PBE incorrectly describe the potential energy surface. They overestimate the redshift of the OH stretching band on hydrogen bond formation which results in an erroneous assignment of the IR bands.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


