Tamalika Ash, Yong Han, James W. Evans and Theresa L. Windus
{"title":"球内水分子对稀土硝酸盐与MCM-22内孔和外表面结合影响的DFT研究","authors":"Tamalika Ash, Yong Han, James W. Evans and Theresa L. Windus","doi":"10.1039/D4CP03424D","DOIUrl":null,"url":null,"abstract":"<p >The impact of inner-sphere water molecules on the binding of rare earth (RE) nitrates to MCM-22 aluminosilicates is analyzed. We used cluster models of MCM-22 to investigate the binding phenomena through localized-basis density functional theory (DFT) calculations. We also conducted plane-wave DFT calculations for a few selected binding configurations using the entire periodic MCM-22 unit cell to check for consistency. Two different MCM-22 cluster models are developed to represent an internal pore and an external surface. Starting with pure silica MCM-22, we substituted one Si with Al and added a H atom on the O bridging the Si and Al to create a Brønsted acid site (BAS), <img>Si–{OH}–Al<img>. Specifically, we investigated the binding of two RE nitrate aqua complexes, [X(NO<small><sub>3</sub></small>)<small><sub>3</sub></small>(H<small><sub>2</sub></small>O)<small><sub>n</sub></small>] where n = 4 (3) for X = Nd (Yb) <em>via</em> the reaction X(NO<small><sub>3</sub></small>)<small><sub>3</sub></small>(H<small><sub>2</sub></small>O)<small><sub>n</sub></small> + <img>Si–{OH}–Al<img> → <img>Si–{OX(NO<small><sub>3</sub></small>)<small><sub>2</sub></small>(H<small><sub>2</sub></small>O)<small><sub>n</sub></small>}–Al<img> + HNO<small><sub>3</sub></small> at BASs, and <em>via</em> an analogous reaction at silanol sites, <img>Si–{OH}. The above analysis just includes the inner coordination sphere H<small><sub>2</sub></small>O. Actually, for the Nd (Yb) complex, after binding at the T1 and T2 sites (T1 site) within the internal pore, one of the H<small><sub>2</sub></small>O molecules leaves this inner sphere. The binding strength at BASs and silanol sites is calculated from the energy change during the above reactions. One finds that Nd complexes prefer binding at the internal pore, while Yb complexes have a comparable binding preference both at the internal pore and external surface. The cluster calculations show good agreement with periodic calculations, implying that the cluster models are suitable for binding studies. Compared to the binding of non-hydrated RE nitrates, the explicit H<small><sub>2</sub></small>O molecules have a minimal impact on overall binding energy trends, but they do increase individual binding energy values. This study also demonstrated the stronger binding affinity of BASs over silanol sites.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 2","pages":" 897-904"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp03424d?page=search","citationCount":"0","resultStr":"{\"title\":\"DFT investigation of the impact of inner-sphere water molecules on RE nitrate binding to internal pore and external surface of MCM-22†\",\"authors\":\"Tamalika Ash, Yong Han, James W. Evans and Theresa L. Windus\",\"doi\":\"10.1039/D4CP03424D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The impact of inner-sphere water molecules on the binding of rare earth (RE) nitrates to MCM-22 aluminosilicates is analyzed. We used cluster models of MCM-22 to investigate the binding phenomena through localized-basis density functional theory (DFT) calculations. We also conducted plane-wave DFT calculations for a few selected binding configurations using the entire periodic MCM-22 unit cell to check for consistency. Two different MCM-22 cluster models are developed to represent an internal pore and an external surface. Starting with pure silica MCM-22, we substituted one Si with Al and added a H atom on the O bridging the Si and Al to create a Brønsted acid site (BAS), <img>Si–{OH}–Al<img>. Specifically, we investigated the binding of two RE nitrate aqua complexes, [X(NO<small><sub>3</sub></small>)<small><sub>3</sub></small>(H<small><sub>2</sub></small>O)<small><sub>n</sub></small>] where n = 4 (3) for X = Nd (Yb) <em>via</em> the reaction X(NO<small><sub>3</sub></small>)<small><sub>3</sub></small>(H<small><sub>2</sub></small>O)<small><sub>n</sub></small> + <img>Si–{OH}–Al<img> → <img>Si–{OX(NO<small><sub>3</sub></small>)<small><sub>2</sub></small>(H<small><sub>2</sub></small>O)<small><sub>n</sub></small>}–Al<img> + HNO<small><sub>3</sub></small> at BASs, and <em>via</em> an analogous reaction at silanol sites, <img>Si–{OH}. The above analysis just includes the inner coordination sphere H<small><sub>2</sub></small>O. Actually, for the Nd (Yb) complex, after binding at the T1 and T2 sites (T1 site) within the internal pore, one of the H<small><sub>2</sub></small>O molecules leaves this inner sphere. The binding strength at BASs and silanol sites is calculated from the energy change during the above reactions. One finds that Nd complexes prefer binding at the internal pore, while Yb complexes have a comparable binding preference both at the internal pore and external surface. The cluster calculations show good agreement with periodic calculations, implying that the cluster models are suitable for binding studies. Compared to the binding of non-hydrated RE nitrates, the explicit H<small><sub>2</sub></small>O molecules have a minimal impact on overall binding energy trends, but they do increase individual binding energy values. This study also demonstrated the stronger binding affinity of BASs over silanol sites.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 2\",\"pages\":\" 897-904\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-12-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp03424d?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03424d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03424d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
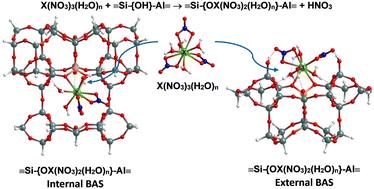
DFT investigation of the impact of inner-sphere water molecules on RE nitrate binding to internal pore and external surface of MCM-22†
The impact of inner-sphere water molecules on the binding of rare earth (RE) nitrates to MCM-22 aluminosilicates is analyzed. We used cluster models of MCM-22 to investigate the binding phenomena through localized-basis density functional theory (DFT) calculations. We also conducted plane-wave DFT calculations for a few selected binding configurations using the entire periodic MCM-22 unit cell to check for consistency. Two different MCM-22 cluster models are developed to represent an internal pore and an external surface. Starting with pure silica MCM-22, we substituted one Si with Al and added a H atom on the O bridging the Si and Al to create a Brønsted acid site (BAS), Si–{OH}–Al. Specifically, we investigated the binding of two RE nitrate aqua complexes, [X(NO3)3(H2O)n] where n = 4 (3) for X = Nd (Yb) via the reaction X(NO3)3(H2O)n + Si–{OH}–Al → Si–{OX(NO3)2(H2O)n}–Al + HNO3 at BASs, and via an analogous reaction at silanol sites, Si–{OH}. The above analysis just includes the inner coordination sphere H2O. Actually, for the Nd (Yb) complex, after binding at the T1 and T2 sites (T1 site) within the internal pore, one of the H2O molecules leaves this inner sphere. The binding strength at BASs and silanol sites is calculated from the energy change during the above reactions. One finds that Nd complexes prefer binding at the internal pore, while Yb complexes have a comparable binding preference both at the internal pore and external surface. The cluster calculations show good agreement with periodic calculations, implying that the cluster models are suitable for binding studies. Compared to the binding of non-hydrated RE nitrates, the explicit H2O molecules have a minimal impact on overall binding energy trends, but they do increase individual binding energy values. This study also demonstrated the stronger binding affinity of BASs over silanol sites.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


