NaCl(100) 上一氧化碳的多维神经网络原子间位势
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
机器学习(ML)模型的出现为原子间势领域带来了新的可能性。我们利用等变图神经网络(NN)构建了一个多功能系统(NaCl(100)表面上的 CO)的原子间势,并以原子动力学的精确度进行了高效的大规模原子模拟。我们报告了两种 NN 电位,一种是在有限温度(T = 30,300 K)下的平衡构型上训练的,另一种是在预激发 CO 吸附物的非平衡轨迹上额外训练的。我们展示了 ML 电位在以下方面的首次应用:(i) 吸附能和反应壁垒;(ii) 亚单层和单层覆盖的势能图;(iii) 有限温度下的振动光谱;(iv) 振动弛豫动力学。还讨论了进一步的可能应用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
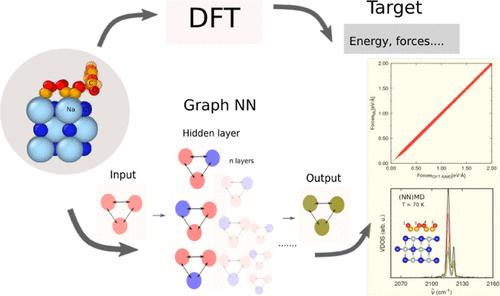
Multidimensional Neural Network Interatomic Potentials for CO on NaCl(100)
The advent of machine learning (ML) models has unlocked new possibilities in the realm of interatomic potentials. We use an equivariant graph Neural Network (NN) to construct interatomic potentials for a versatile system, CO on a NaCl(100) surface, and mediate efficient large-scale atomistic simulations with ab initio molecular dynamics accuracy. We report two NN potentials, one trained on equilibrium configurations at finite temperatures (T = 30, 300 K), and the other additionally trained upon nonequilibrium trajectories of pre-excited CO adsorbates. We demonstrate first applications of the ML potentials for (i) adsorption energies and barriers for reactions, (ii) potential energy landscapes for submonolayer and monolayer coverages, (iii) vibrational spectra at finite temperatures, and (iv) vibrational relaxation dynamics. Further possible applications are discussed.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


