新型 Au3 簇装饰砷烯纳米片对噻吩和甲硫醇检测的吸附行为研究:DFT 研究
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
第一性原理计算描述了 Aun-arsenene 纳米片与甲硫醇/噻吩分子之间的相互作用。由于在费米水平附近出现了新的能带,Au-砒霜烯和 Au3-砒霜烯纳米片都显示出比纯砒霜烯系统更小的带隙。因此,金功能化砷烯纳米片的电导率和传感器功能显著增强。Au-arsenene 和 Au3-arsenene 纳米片的计算形成能分别为 -2.38 eV 和 -5.37 eV,这表明它们具有稳定的结构。由于计算得出的吸附能为负值,甲硫醇和噻吩分子会自发地吸附在金-烯烃基底上。此外,噻吩分子通过其与金原子紧密相连的 S 原子在金-烯基底上显示出最高的吸附能(-0.52 eV)。适中的吸附能表明传感应用的恢复时间更短。我们的工作为探索金-烯烃作为传感甲硫醇和噻吩分子的有效候选材料提供了启示。本文章由计算机程序翻译,如有差异,请以英文原文为准。
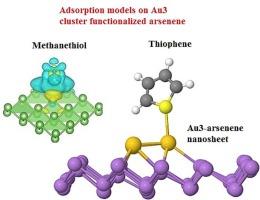
Investigation of the adsorption behaviors of novel Au3 cluster decorated arsenene nanosheets towards thiophene and methanethiol detection: A DFT study
First principles calculations were carried out to describe the interactions between Aun-arsenene nanosheets and methanethiol/thiophene molecules. Both Au-arsenene and Au3-arsenene nanosheets exhibit smaller band gap than the pure arsenene system due to the emergence of new energy bands near the Fermi level. Thus, the conductivity and sensor capabilities are significantly enhanced for the Au functionalized arsenene nanosheets. Calculated formation energies of −2.38 eV and −5.37 eV for Au-arsenene and Au3-arsenene nanosheets indicate their structural stability. The adsorption of methanethiol and thiophene molecules on the Au-arsenene substrates spontaneously occurs due to the negative calculated adsorption energies. Besides, thiophene molecule exhibits the highest adsorption energy (−0.52 eV) on the Au-arsenene substrate through its S atom strongly connected to the Au atom. The moderate adsorption energies indicate the shorter recovery times for sensing applications. Our work sheds light on the exploration of Au-arsenene as an effective candidate for sensing methanethiol and thiophene molecules.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


