{"title":"2005-2019年中国东南部福建省志贺氏菌临床分离株的基因组和抗生素耐药性特征。","authors":"Mengying Huang, Xiaoxuan Zhang, Chaochen Luo, Haibin Xu, Yufeng Qiu, Jinsong Yang","doi":"10.1099/mgen.0.001325","DOIUrl":null,"url":null,"abstract":"<p><p>Shigellosis is a serious public health issue in many developing countries. The emergence of multidrug-resistant (MDR) <i>Shigella</i> isolates has deepened the treatment difficulty and health burden of shigellosis. China is the largest developing country in the world, but so far, the genome of MDR <i>Shigella</i> isolates has not been well characterized. In this study, 60 clinical isolates of <i>Shigella</i> spp. in Fujian Province, southeast China, from 2005 to 2019 were characterized for drug resistance phenotype, whole-genome sequencing and bioinformatics analysis. The results showed that the MDR rate of <i>Shigella</i> isolates was 100%, among which the resistance rates of cefotaxime, ciprofloxacin and azithromycin were 36.67, 21.67 and 10.00 %, respectively. The positive rate of extended-spectrum beta-lactamase (ESBL)-producing strains was 23.33%. The resistance profiles of <i>Shigella flexneri</i> and <i>Shigella sonnei</i> to some antimicrobials differed. The MDR isolates carried multiple antimicrobial resistance genes, among which <i>blaCTX-M-14</i> and <i>blaCTX-M-15</i> mediated ESBL resistance<i>.</i> '<i>ISEcp1 -blaCTX-M -IS903</i>' (type I) and '<i>ISEcp1 -blaCTX-M</i>' (type II) were the most common genetic environments around the <i>blaCTX-M</i> genes, and plasmids containing these structures included IncFII, IncI1, IncI2 and IncN. The double gene mutation pattern of <i>gyr</i>A and <i>par</i>C resulted in a significant decrease in the sensitivity of <i>S. flexneri</i> to ciprofloxacin. The overall resistance phenotype and genotype concordance rate was 88.50%, and the sensitivity and specificity of the genotype antimicrobial susceptibility test (AST) were 93.35 and 82.53 %, respectively. However, inconsistency occurred between phenotypic and genotype profiles for a few antibiotics. Phylogenomic investigation with core genome multi-locus sequence typing (cgMLST) and SNPs were used to characterize the endemic transmission of these infections in Fujian and their evolutionary origin within the global context. For <i>S. flexneri</i>, Fujian isolates were all limited to PG3 and could be divided into three phylogenetic clusters. The ciprofloxacin-resistant strains were mainly F2a and FXv and assigned to the three clusters with different quinolone resistance-determining region mutation patterns. For <i>S. sonnei</i>, most Fujian strains belonged to Lineage III with genotype 3.7.6, except three isolates of Lineage I with genotype 1.3. The strains carrying the <i>blaCTX-M</i> genes were dispersed, indicating different origins of gene acquisition. Most of the circulating isolates in Fujian Province were not related to major international outbreak lineages and were only endemic to the country. In conclusion, multi-drug resistance of <i>Shigella</i> isolates in Fujian Province was serious, and genome-based laboratory surveillance will be crucial to the clinical treatment and public health measures for shigellosis.</p>","PeriodicalId":18487,"journal":{"name":"Microbial Genomics","volume":"10 11","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11893363/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genome and antibiotic resistance characteristics of <i>Shigella</i> clinical isolates in Fujian Province, Southeast China, 2005-2019.\",\"authors\":\"Mengying Huang, Xiaoxuan Zhang, Chaochen Luo, Haibin Xu, Yufeng Qiu, Jinsong Yang\",\"doi\":\"10.1099/mgen.0.001325\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Shigellosis is a serious public health issue in many developing countries. The emergence of multidrug-resistant (MDR) <i>Shigella</i> isolates has deepened the treatment difficulty and health burden of shigellosis. China is the largest developing country in the world, but so far, the genome of MDR <i>Shigella</i> isolates has not been well characterized. In this study, 60 clinical isolates of <i>Shigella</i> spp. in Fujian Province, southeast China, from 2005 to 2019 were characterized for drug resistance phenotype, whole-genome sequencing and bioinformatics analysis. The results showed that the MDR rate of <i>Shigella</i> isolates was 100%, among which the resistance rates of cefotaxime, ciprofloxacin and azithromycin were 36.67, 21.67 and 10.00 %, respectively. The positive rate of extended-spectrum beta-lactamase (ESBL)-producing strains was 23.33%. The resistance profiles of <i>Shigella flexneri</i> and <i>Shigella sonnei</i> to some antimicrobials differed. The MDR isolates carried multiple antimicrobial resistance genes, among which <i>blaCTX-M-14</i> and <i>blaCTX-M-15</i> mediated ESBL resistance<i>.</i> '<i>ISEcp1 -blaCTX-M -IS903</i>' (type I) and '<i>ISEcp1 -blaCTX-M</i>' (type II) were the most common genetic environments around the <i>blaCTX-M</i> genes, and plasmids containing these structures included IncFII, IncI1, IncI2 and IncN. The double gene mutation pattern of <i>gyr</i>A and <i>par</i>C resulted in a significant decrease in the sensitivity of <i>S. flexneri</i> to ciprofloxacin. The overall resistance phenotype and genotype concordance rate was 88.50%, and the sensitivity and specificity of the genotype antimicrobial susceptibility test (AST) were 93.35 and 82.53 %, respectively. However, inconsistency occurred between phenotypic and genotype profiles for a few antibiotics. Phylogenomic investigation with core genome multi-locus sequence typing (cgMLST) and SNPs were used to characterize the endemic transmission of these infections in Fujian and their evolutionary origin within the global context. For <i>S. flexneri</i>, Fujian isolates were all limited to PG3 and could be divided into three phylogenetic clusters. The ciprofloxacin-resistant strains were mainly F2a and FXv and assigned to the three clusters with different quinolone resistance-determining region mutation patterns. For <i>S. sonnei</i>, most Fujian strains belonged to Lineage III with genotype 3.7.6, except three isolates of Lineage I with genotype 1.3. The strains carrying the <i>blaCTX-M</i> genes were dispersed, indicating different origins of gene acquisition. Most of the circulating isolates in Fujian Province were not related to major international outbreak lineages and were only endemic to the country. In conclusion, multi-drug resistance of <i>Shigella</i> isolates in Fujian Province was serious, and genome-based laboratory surveillance will be crucial to the clinical treatment and public health measures for shigellosis.</p>\",\"PeriodicalId\":18487,\"journal\":{\"name\":\"Microbial Genomics\",\"volume\":\"10 11\",\"pages\":\"\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11893363/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microbial Genomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1099/mgen.0.001325\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1099/mgen.0.001325","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genome and antibiotic resistance characteristics of Shigella clinical isolates in Fujian Province, Southeast China, 2005-2019.
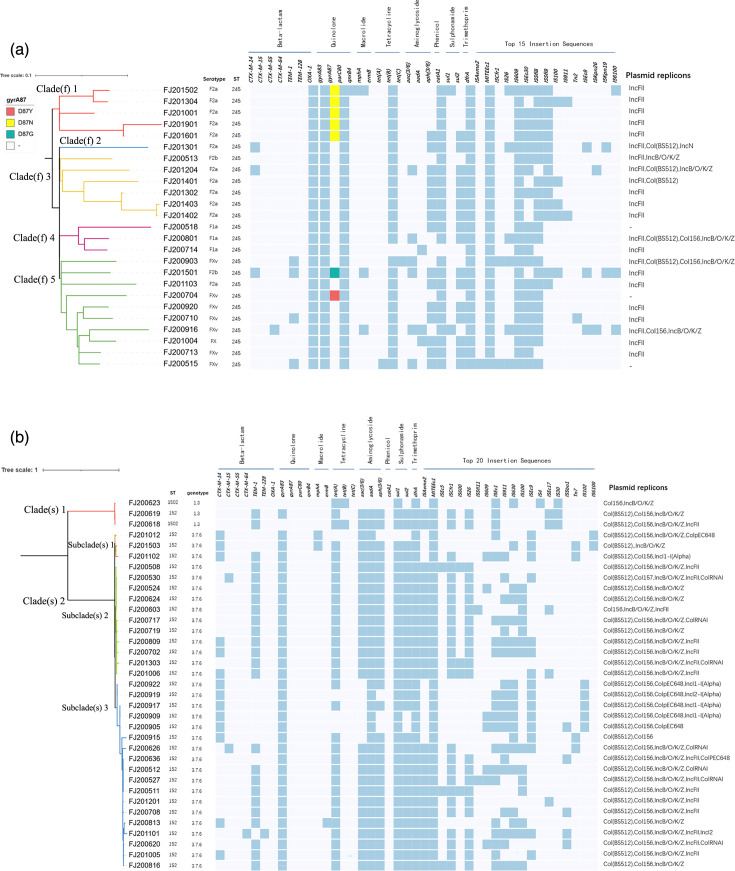
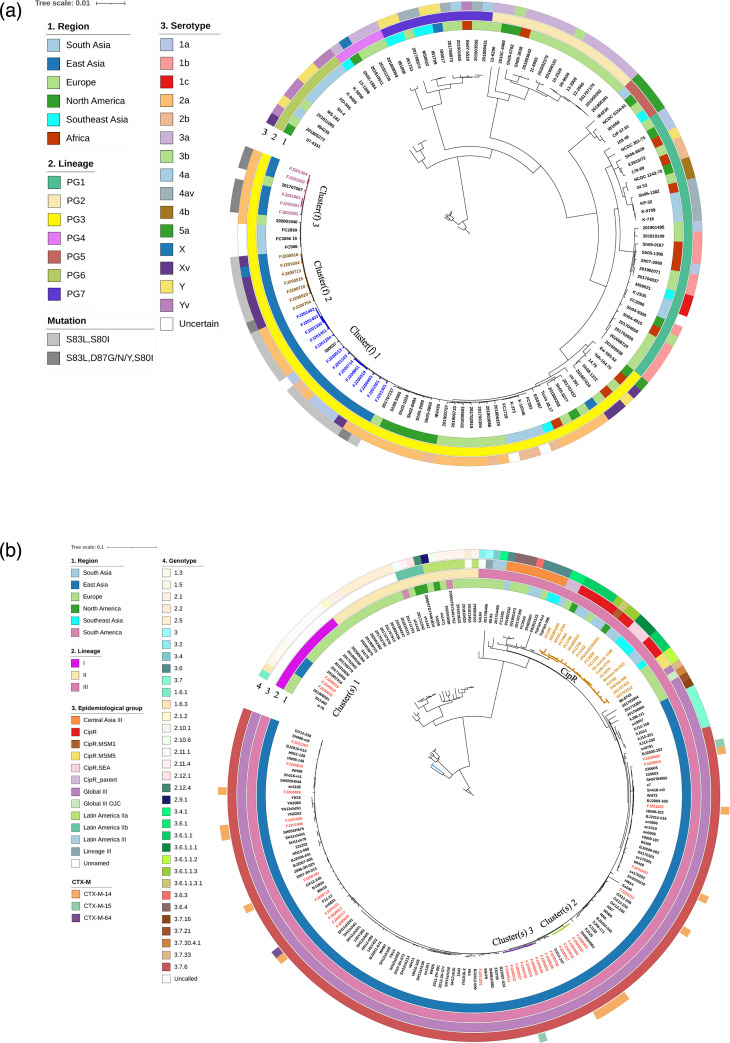
Shigellosis is a serious public health issue in many developing countries. The emergence of multidrug-resistant (MDR) Shigella isolates has deepened the treatment difficulty and health burden of shigellosis. China is the largest developing country in the world, but so far, the genome of MDR Shigella isolates has not been well characterized. In this study, 60 clinical isolates of Shigella spp. in Fujian Province, southeast China, from 2005 to 2019 were characterized for drug resistance phenotype, whole-genome sequencing and bioinformatics analysis. The results showed that the MDR rate of Shigella isolates was 100%, among which the resistance rates of cefotaxime, ciprofloxacin and azithromycin were 36.67, 21.67 and 10.00 %, respectively. The positive rate of extended-spectrum beta-lactamase (ESBL)-producing strains was 23.33%. The resistance profiles of Shigella flexneri and Shigella sonnei to some antimicrobials differed. The MDR isolates carried multiple antimicrobial resistance genes, among which blaCTX-M-14 and blaCTX-M-15 mediated ESBL resistance. 'ISEcp1 -blaCTX-M -IS903' (type I) and 'ISEcp1 -blaCTX-M' (type II) were the most common genetic environments around the blaCTX-M genes, and plasmids containing these structures included IncFII, IncI1, IncI2 and IncN. The double gene mutation pattern of gyrA and parC resulted in a significant decrease in the sensitivity of S. flexneri to ciprofloxacin. The overall resistance phenotype and genotype concordance rate was 88.50%, and the sensitivity and specificity of the genotype antimicrobial susceptibility test (AST) were 93.35 and 82.53 %, respectively. However, inconsistency occurred between phenotypic and genotype profiles for a few antibiotics. Phylogenomic investigation with core genome multi-locus sequence typing (cgMLST) and SNPs were used to characterize the endemic transmission of these infections in Fujian and their evolutionary origin within the global context. For S. flexneri, Fujian isolates were all limited to PG3 and could be divided into three phylogenetic clusters. The ciprofloxacin-resistant strains were mainly F2a and FXv and assigned to the three clusters with different quinolone resistance-determining region mutation patterns. For S. sonnei, most Fujian strains belonged to Lineage III with genotype 3.7.6, except three isolates of Lineage I with genotype 1.3. The strains carrying the blaCTX-M genes were dispersed, indicating different origins of gene acquisition. Most of the circulating isolates in Fujian Province were not related to major international outbreak lineages and were only endemic to the country. In conclusion, multi-drug resistance of Shigella isolates in Fujian Province was serious, and genome-based laboratory surveillance will be crucial to the clinical treatment and public health measures for shigellosis.
期刊介绍:
Microbial Genomics (MGen) is a fully open access, mandatory open data and peer-reviewed journal publishing high-profile original research on archaea, bacteria, microbial eukaryotes and viruses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


