{"title":"RiceSNP-BST:预测水稻生物胁迫相关 SNP 的深度学习框架。","authors":"Jiajun Xu, Yujia Gao, Quan Lu, Renyi Zhang, Jianfeng Gui, Xiaoshuang Liu, Zhenyu Yue","doi":"10.1093/bib/bbae599","DOIUrl":null,"url":null,"abstract":"<p><p>Rice consistently faces significant threats from biotic stresses, such as fungi, bacteria, pests, and viruses. Consequently, accurately and rapidly identifying previously unknown single-nucleotide polymorphisms (SNPs) in the rice genome is a critical challenge for rice research and the development of resistant varieties. However, the limited availability of high-quality rice genotype data has hindered this research. Deep learning has transformed biological research by facilitating the prediction and analysis of SNPs in biological sequence data. Convolutional neural networks are especially effective in extracting structural and local features from DNA sequences, leading to significant advancements in genomics. Nevertheless, the expanding catalog of genome-wide association studies provides valuable biological insights for rice research. Expanding on this idea, we introduce RiceSNP-BST, an automatic architecture search framework designed to predict SNPs associated with rice biotic stress traits (BST-associated SNPs) by integrating multidimensional features. Notably, the model successfully innovates the datasets, offering more precision than state-of-the-art methods while demonstrating good performance on an independent test set and cross-species datasets. Additionally, we extracted features from the original DNA sequences and employed causal inference to enhance the biological interpretability of the model. This study highlights the potential of RiceSNP-BST in advancing genome prediction in rice. Furthermore, a user-friendly web server for RiceSNP-BST (http://rice-snp-bst.aielab.cc) has been developed to support broader genome research.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"25 6","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11576077/pdf/","citationCount":"0","resultStr":"{\"title\":\"RiceSNP-BST: a deep learning framework for predicting biotic stress-associated SNPs in rice.\",\"authors\":\"Jiajun Xu, Yujia Gao, Quan Lu, Renyi Zhang, Jianfeng Gui, Xiaoshuang Liu, Zhenyu Yue\",\"doi\":\"10.1093/bib/bbae599\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Rice consistently faces significant threats from biotic stresses, such as fungi, bacteria, pests, and viruses. Consequently, accurately and rapidly identifying previously unknown single-nucleotide polymorphisms (SNPs) in the rice genome is a critical challenge for rice research and the development of resistant varieties. However, the limited availability of high-quality rice genotype data has hindered this research. Deep learning has transformed biological research by facilitating the prediction and analysis of SNPs in biological sequence data. Convolutional neural networks are especially effective in extracting structural and local features from DNA sequences, leading to significant advancements in genomics. Nevertheless, the expanding catalog of genome-wide association studies provides valuable biological insights for rice research. Expanding on this idea, we introduce RiceSNP-BST, an automatic architecture search framework designed to predict SNPs associated with rice biotic stress traits (BST-associated SNPs) by integrating multidimensional features. Notably, the model successfully innovates the datasets, offering more precision than state-of-the-art methods while demonstrating good performance on an independent test set and cross-species datasets. Additionally, we extracted features from the original DNA sequences and employed causal inference to enhance the biological interpretability of the model. This study highlights the potential of RiceSNP-BST in advancing genome prediction in rice. Furthermore, a user-friendly web server for RiceSNP-BST (http://rice-snp-bst.aielab.cc) has been developed to support broader genome research.</p>\",\"PeriodicalId\":9209,\"journal\":{\"name\":\"Briefings in bioinformatics\",\"volume\":\"25 6\",\"pages\":\"\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2024-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11576077/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Briefings in bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bib/bbae599\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbae599","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
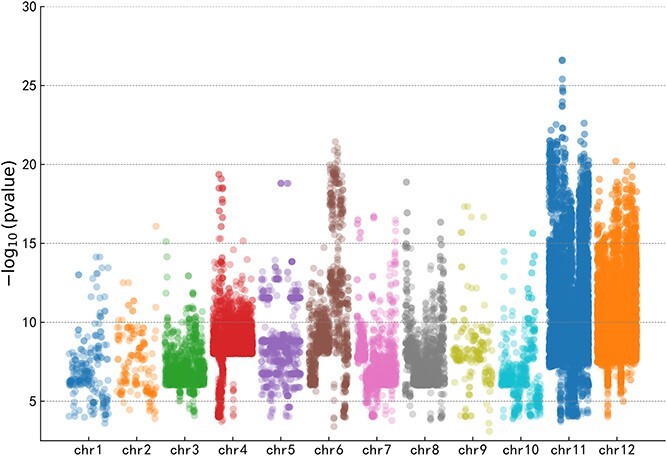
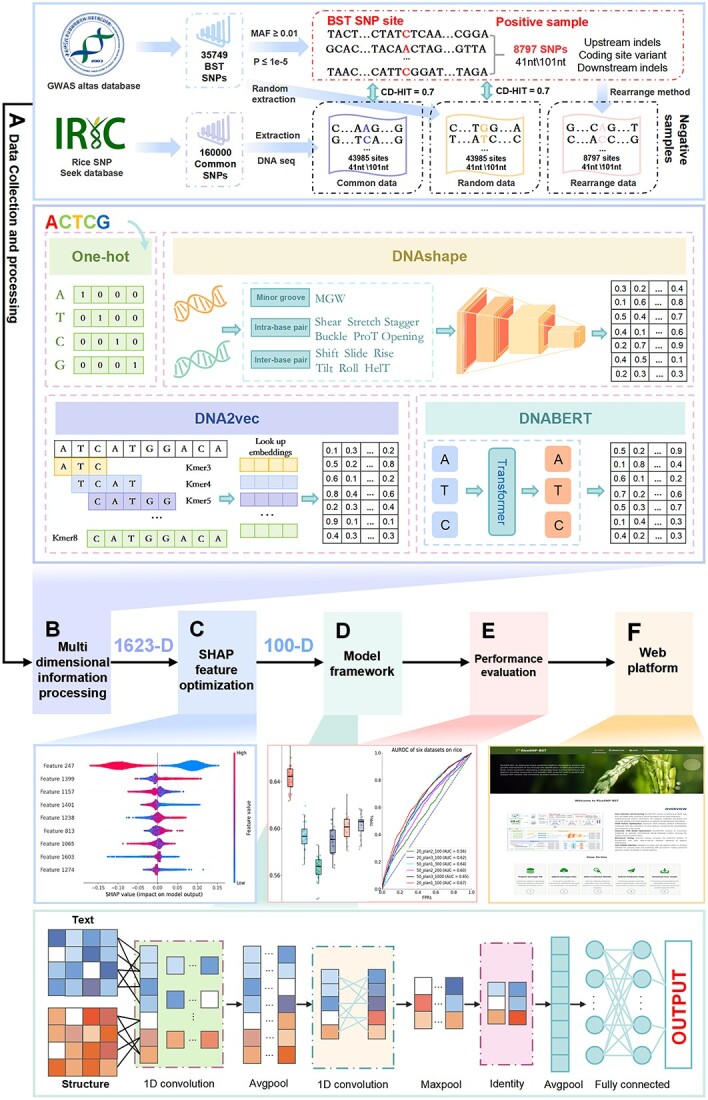
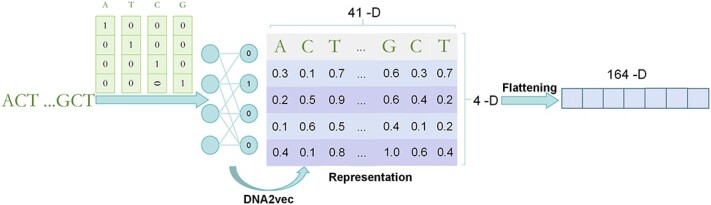
水稻一直面临着真菌、细菌、害虫和病毒等生物胁迫的严重威胁。因此,准确、快速地鉴定水稻基因组中先前未知的单核苷酸多态性(SNPs)是水稻研究和抗病品种开发面临的关键挑战。然而,高质量水稻基因型数据的有限可用性阻碍了这项研究。深度学习促进了生物序列数据中 SNP 的预测和分析,从而改变了生物学研究。卷积神经网络在从 DNA 序列中提取结构和局部特征方面尤为有效,从而在基因组学领域取得了重大进展。然而,不断扩大的全基因组关联研究为水稻研究提供了宝贵的生物学见解。基于这一想法,我们引入了 RiceSNP-BST,这是一个自动结构搜索框架,旨在通过整合多维特征来预测与水稻生物胁迫性状相关的 SNPs(BST 相关 SNPs)。值得注意的是,该模型成功地对数据集进行了创新,与最先进的方法相比精度更高,同时在独立测试集和跨物种数据集上表现出良好的性能。此外,我们还从原始 DNA 序列中提取了特征,并采用因果推理来增强模型的生物学可解释性。这项研究凸显了 RiceSNP-BST 在推进水稻基因组预测方面的潜力。此外,我们还为 RiceSNP-BST 开发了一个用户友好型网络服务器 (http://rice-snp-bst.aielab.cc),以支持更广泛的基因组研究。
RiceSNP-BST: a deep learning framework for predicting biotic stress-associated SNPs in rice.
Rice consistently faces significant threats from biotic stresses, such as fungi, bacteria, pests, and viruses. Consequently, accurately and rapidly identifying previously unknown single-nucleotide polymorphisms (SNPs) in the rice genome is a critical challenge for rice research and the development of resistant varieties. However, the limited availability of high-quality rice genotype data has hindered this research. Deep learning has transformed biological research by facilitating the prediction and analysis of SNPs in biological sequence data. Convolutional neural networks are especially effective in extracting structural and local features from DNA sequences, leading to significant advancements in genomics. Nevertheless, the expanding catalog of genome-wide association studies provides valuable biological insights for rice research. Expanding on this idea, we introduce RiceSNP-BST, an automatic architecture search framework designed to predict SNPs associated with rice biotic stress traits (BST-associated SNPs) by integrating multidimensional features. Notably, the model successfully innovates the datasets, offering more precision than state-of-the-art methods while demonstrating good performance on an independent test set and cross-species datasets. Additionally, we extracted features from the original DNA sequences and employed causal inference to enhance the biological interpretability of the model. This study highlights the potential of RiceSNP-BST in advancing genome prediction in rice. Furthermore, a user-friendly web server for RiceSNP-BST (http://rice-snp-bst.aielab.cc) has been developed to support broader genome research.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


