{"title":"3,5 二甲基异噁唑通过 S1 电子态发生光异构化反应的非绝热 ab initio 化学反应动力学","authors":"Mizuki Kimura and Shinkoh Nanbu","doi":"10.1039/D4CP03137G","DOIUrl":null,"url":null,"abstract":"<p >Nonadiabatic <em>ab initio</em> molecular dynamics simulations were performed to explore the photoisomerization pathway from isoxazole (iso-OXA) to oxazole (OXA), considering four electronic states. The XMS-CASPT2 and SA4-CASSCF theories were employed to describe these electronic structures, which were caused by 12 electrons in 11 orbitals with the cc-pVDZ + sp diffuse basis set; the Gaussian s- and p-type diffuse functions were extracted from Dunning's aug-cc-pVDZ function. The potential energy and its gradient at each time step were computed on-the-fly at these levels in the time evolution of the classical trajectory. When the two electronic states were close to each other, the trajectory surface hopping (TSH) judgment between the two adjacent states was carried out by the anteater procedure based on the Zhu–Nakamura formula (ZN-TSH). The two different excited state lifetimes were found to exist in the first electronic state (S<small><sub>1</sub></small>), estimated at 10.77 and 119.81 fs. Upon photoexcitation, the N–O bond breaks and energetically relaxes to the ground state (S<small><sub>0</sub></small>). In the pathway leading to the main product, azirine formation, the 5-membered ring retains a planar structure while undergoing a non-adiabatic transition with an increasing N–O bond distance. Furthermore, it was verified that a 1,2-shift takes place in the pathway that results in the production of ketenimine, causing a nonadiabatic transition.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 1","pages":" 62-76"},"PeriodicalIF":2.9000,"publicationDate":"2024-11-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp03137g?page=search","citationCount":"0","resultStr":"{\"title\":\"Nonadiabatic ab initio chemical reaction dynamics for the photoisomerization reaction of 3,5-dimethylisoxazole via the S1 electronic state†\",\"authors\":\"Mizuki Kimura and Shinkoh Nanbu\",\"doi\":\"10.1039/D4CP03137G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Nonadiabatic <em>ab initio</em> molecular dynamics simulations were performed to explore the photoisomerization pathway from isoxazole (iso-OXA) to oxazole (OXA), considering four electronic states. The XMS-CASPT2 and SA4-CASSCF theories were employed to describe these electronic structures, which were caused by 12 electrons in 11 orbitals with the cc-pVDZ + sp diffuse basis set; the Gaussian s- and p-type diffuse functions were extracted from Dunning's aug-cc-pVDZ function. The potential energy and its gradient at each time step were computed on-the-fly at these levels in the time evolution of the classical trajectory. When the two electronic states were close to each other, the trajectory surface hopping (TSH) judgment between the two adjacent states was carried out by the anteater procedure based on the Zhu–Nakamura formula (ZN-TSH). The two different excited state lifetimes were found to exist in the first electronic state (S<small><sub>1</sub></small>), estimated at 10.77 and 119.81 fs. Upon photoexcitation, the N–O bond breaks and energetically relaxes to the ground state (S<small><sub>0</sub></small>). In the pathway leading to the main product, azirine formation, the 5-membered ring retains a planar structure while undergoing a non-adiabatic transition with an increasing N–O bond distance. Furthermore, it was verified that a 1,2-shift takes place in the pathway that results in the production of ketenimine, causing a nonadiabatic transition.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 1\",\"pages\":\" 62-76\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-11-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp03137g?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03137g\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03137g","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
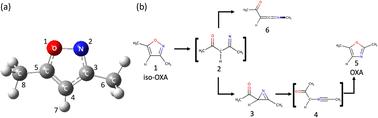
为了探索从异噁唑(iso-OXA)到噁唑(OXA)的光异构化途径,我们进行了非绝热 ab initio 分子动力学模拟,并考虑了四种电子状态。采用 XMS-CASPT2 和 SA4-CASSCF 理论来描述这些电子结构,它们是由 11 个轨道上的 12 个电子和 cc-pVDZ + sp 扩散基集引起的;高斯 s 型和 p 型扩散函数是从 Dunning 的 aug-cc-pVDZ 函数中提取的。每个时间步的势能及其梯度是在经典轨迹的时间演化过程中在这些水平上即时计算的。当两个电子态彼此接近时,根据朱-中村公式(ZN-TSH)的蚂蚁程序在两个相邻态之间进行轨迹表面跳跃(TSH)判断。发现第一电子态(S1)存在两种不同的激发态寿命,估计分别为 10.77 和 119.81 fs。光激发时,N-O 键断裂并能量弛豫到基态(S0)。在形成主要产物氮丙啶的过程中,五元环保持平面结构,同时随着 N-O 键距离的增加而发生非绝热转变。此外,还验证了在生成酮亚胺的途径中发生了 1,2 转变,导致非绝热转变。
Nonadiabatic ab initio chemical reaction dynamics for the photoisomerization reaction of 3,5-dimethylisoxazole via the S1 electronic state†
Nonadiabatic ab initio molecular dynamics simulations were performed to explore the photoisomerization pathway from isoxazole (iso-OXA) to oxazole (OXA), considering four electronic states. The XMS-CASPT2 and SA4-CASSCF theories were employed to describe these electronic structures, which were caused by 12 electrons in 11 orbitals with the cc-pVDZ + sp diffuse basis set; the Gaussian s- and p-type diffuse functions were extracted from Dunning's aug-cc-pVDZ function. The potential energy and its gradient at each time step were computed on-the-fly at these levels in the time evolution of the classical trajectory. When the two electronic states were close to each other, the trajectory surface hopping (TSH) judgment between the two adjacent states was carried out by the anteater procedure based on the Zhu–Nakamura formula (ZN-TSH). The two different excited state lifetimes were found to exist in the first electronic state (S1), estimated at 10.77 and 119.81 fs. Upon photoexcitation, the N–O bond breaks and energetically relaxes to the ground state (S0). In the pathway leading to the main product, azirine formation, the 5-membered ring retains a planar structure while undergoing a non-adiabatic transition with an increasing N–O bond distance. Furthermore, it was verified that a 1,2-shift takes place in the pathway that results in the production of ketenimine, causing a nonadiabatic transition.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


