钌(II)配合物催化的光催化底物氧化作用(以吩嗪分子为活性位点,以二氧为末端氧化剂
IF 15.6
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
我们开发了以 O2 为末端氧化剂对芳香族底物进行光催化氧化的方法,在可见光照射下,在酸性水中只生成 2e 氧化产物,而无需对 O2 进行还原活化。一种以四吡啶并[3,2-a:2′,3′-c:3″,2″-h:2‴,3‴-j]吩嗪(tpphz)为配体、以吡嗪分子为活性位点的 RuII 复合物(1)被用作光催化剂。光催化的活性物种不是复合物 1 本身,而是在 tpphz 的二亚胺空位上质子化的质子化形式 1-H+。在有机底物存在下进行光激发时,1-H+ 转化为相应的二氢中间体(2-H+),配体的吡嗪基从底物中获得 2e- 和 2H+。2-H+ 很容易被 O2 氧化,从而回收 1-H+。因此,得到了底物的氧化产物和二氧还原产生的 H2O2;不过,形成的 H2O2 也被用于氧化 2-H+。在苯甲醇氧化成苯甲醛的过程中,10 小时的周转次数达到 240,量子产率为 4.0%。环丁醇氧化过程中没有开环产物,这表明催化反应是通过涉及形式氢化物转移的机理进行的。机理研究表明,1-H+ 对底物的光催化氧化既不是在最低单激发态也不是在三重激发态(S1 或 T1)实现的,而是在第二低单激发态(S2),即 tpphz 配体的 1(π-π*)*。因此,1-H+ 对底物的光催化氧化作用可归类为不寻常的反卡沙光催化作用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
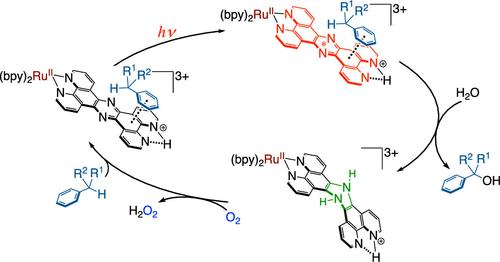
Photocatalytic Substrate Oxidation Catalyzed by a Ruthenium(II) Complex with a Phenazine Moiety as the Active Site Using Dioxygen as a Terminal Oxidant
We have developed photocatalytic oxidation of aromatic substrates using O2 as a terminal oxidant to afford only 2e–-oxidized products without the reductive activation of O2 in acidic water under visible-light irradiation. A RuII complex (1) bearing a pyrazine moiety as the active site in tetrapyrido[3,2-a:2′,3′-c:3″,2″-h:2‴,3‴-j]phenazine (tpphz) as a ligand was employed as a photocatalyst. The active species for the photocatalysis was revealed to be not complex 1 itself but the protonated form, 1-H+, protonated at the vacant diimine site of tpphz. Upon photoexcitation in the presence of an organic substrate, 1-H+ was converted to the corresponding dihydro-intermediate (2-H+), where the pyrazine moiety of the ligand received 2e– and 2H+ from the substrate. 2-H+ was facilely oxidized by O2 to recover 1-H+. Consequently, an oxidation product of the substrate and H2O2 derived from dioxygen reduction were obtained; however, the H2O2 formed was also used for oxidation of 2-H+. In the oxidation of benzyl alcohol to benzaldehyde, the turnover number reached 240 for 10 h, and the quantum yield was determined to be 4.0%. The absence of ring-opening products in the oxidation of cyclobutanol suggests that the catalytic reaction proceeds through a mechanism involving formal hydride transfer. Mechanistic studies revealed that the photocatalytic substrate oxidation by 1-H+ was achieved in neither the lowest singlet excited state nor triplet excited state (S1 or T1) but in the second lowest singlet excited state (S2), i.e., 1(π–π*)* of the tpphz ligand. Thus, the photocatalytic substrate oxidation by 1-H+ can be categorized into unusual anti-Kasha photocatalysis.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


