Carina S. T. Peraça, Karla F. Andriani, Maurício J. Piotrowski, Juarez L. F. Da Silva
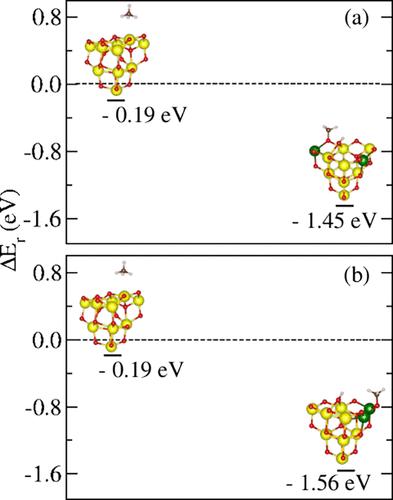
{"title":"对 \"CH4 在 (CeO2)10 簇上脱氢的 Ab Initio 研究 \"的更正","authors":"Carina S. T. Peraça, Karla F. Andriani, Maurício J. Piotrowski, Juarez L. F. Da Silva","doi":"10.1021/acs.jpcc.4c07452","DOIUrl":null,"url":null,"abstract":"Unfortunately, we identified two errors in our paper related to the misapplication of the unity bond index–quadratic exponential potential (UBI–QEP) analysis, which became evident through discussions with Dr. Verónica Ganduglia-Pirovano and Dr. Breno L. Galvão (Private communication, March 15, 2024). The first error involves the results used to design Figure 5, where we obtained the activation energy values using the UBI-QEP approximation. In this analysis, we considered the most common adsorption sites reported in the literature for the CeO<sub>2</sub>(111) surface rather than the most stable ones on the ceria cluster. As a result, the dependence between the CH<sub>4</sub> adsorption site and the corresponding CH<sub>3</sub> and/or H sites was not maintained. A more appropriate approach for our purposes would be to conduct a systematic investigation of the coadsorption sites on (CeO<sub>2</sub>)<sub>10</sub>, using a specific adsorption site as a reference and mapping the most stable CH<sub>3</sub> or H sites. Therefore, we implemented the aforementioned approach by mapping the CH<sub>3</sub> sites and fixing the most stable H site as a reference; hence, the text and the related figure (Figure 5) on page 11944 should be updated based on the obtained new results. The second error concerns the formulation of Figure 6, where the transverse lines connecting the structures may give the reader the mistaken impression of a proposed reaction path. In reality, we present the reaction energy values for representative structures of each group selected by the <i>k-means</i> clustering method. Regarding the mistake in the production of Figures 5 and 6, the following changes must be applied: On page 11946, the sentence “The CH<sub>4</sub> molecule can adsorb on the nanocluster considering four adsorption configurations, [...]” must be replaced with “The CH<sub>4</sub> molecule can adsorb on the nanocluster considering three adsorption configurations, [...]” and the sentence “[...] C–H bond break on the (CeO<sub>2</sub>)<sub>10</sub> nanocluster can be related to the H adsorption site [...]” must be replaced with “[...] C–H bond break on the (CeO<sub>2</sub>)<sub>10</sub> nanocluster can be related to the coadsorption sites of CH<sub>3</sub> (H)[...]”. The original Figure 5 should be replaced by Figure 5 of this Correction. On page 11944, the sentence “We found four different adsorption modes for CH<sub>4</sub> on (CeO<sub>2</sub>)<sub>10</sub>, namely, umbrella, antiumbrella, scissoring, and modified antiumbrella, as indicated in Figure 5.<sup>66</sup> In the umbrella and antiumbrella modes, the calculated barrier (<i>E</i><sub>a</sub>) for the CH<sub>4</sub> dissociation (CH<sub>4</sub> → CH<sub>3</sub> + H*) indicates a higher cluster reactivity compared to the CeO<sub>2</sub>(111) surface. In this case, we obtained <i>E</i><sub>a</sub> equal to 0.54 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.48 eV) and 0.59 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.52 eV) for umbrella and antiumbrella modes, respectively, smaller than the value reported for the CeO<sub>2</sub>(111) surface (1.44 eV).<sup>65</sup>” must be replaced with “We found three different adsorption modes for CH<sub>4</sub> on (CeO<sub>2</sub>)<sub>10</sub> clusters, namely umbrella, antiumbrella, and scissoring.<sup>66</sup> The calculated barrier (<i>E</i><sub>a</sub>) for the CH<sub>4</sub> dissociation (CH<sub>4</sub> → CH<sub>3</sub> + H*) indicates a higher cluster reactivity compared to the CeO<sub>2</sub>(111) surface. In this case, we obtained <i>E</i><sub>a</sub> equal to 0.07 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.03) and 0.13 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.09 eV) for the configuration proposed in Figure 1a,b, respectively, smaller than the value reported for the CeO<sub>2</sub>(111) surface (1.44 eV).<sup>65</sup>” In page 11944, the sentence “Then, the adsorption energy for the umbrella and antiumbrella modes in this state is −3.59 and −3.95 eV, respectively, in agreement with the value of −3.68 eV reported for CeO<sub>2</sub>(111) surface.<sup>25</sup>” must be removed. The original Figure 6 should be replaced by Figure 6 of this Correction. On page 11945, the following paragraph must be removed: “The energy barriers for the modified antiumbrella and scissoring modes (0.81 and 0.64 eV, respectively) indicate a higher barrier compared to the umbrella and antiumbrella modes, which can be associated with the H adsorption site. Once again, the ZPE corrections do not produce significant changes in the <i>E</i><sub>a</sub> values (0.74 and 0.56 eV, respectively). The difference between the modified and antiumbrella modes is the H adsorption site, since the former adsorbs in the 3-fold top<sup>O</sup> site, allowing the molecule adsorption at a 2-fold top<sup>O</sup> site. The opposite occurs for the latter one, in which the molecule adsorbs on the adsorption O site with a higher coordination. However, the activation energy obtained for the modified antiumbrella and scissoring mode can be compared with the values reported by Righi et al. for pristine CeO<sub>2</sub>(100) surface (0.87 eV).<sup>25</sup>” Table S48 has been corrected in the Supporting Information file and replaced by Table S48 here. The corrections mentioned do not affect the other discussions or our general conclusions. We regret any inconveniences. The authors thank Verónica Ganduglia–Pirovano and Breno L. Galvão for their valuable scientific discussions.<named-content content-type=\"anchor\" r type=\"simple\"></named-content><named-content content-type=\"anchor\" r type=\"simple\"></named-content><named-content content-type=\"anchor\" r type=\"simple\"></named-content> Figure 5. Relative reaction energy (Δ<i>E</i><sub>r</sub>) for the CH<sub>4</sub> → CH<sub>3</sub> + H step of CH<sub>4</sub> dehydrogenation. Frames (a) and (b) represent the two most stable (CH<sub>3</sub> + H)/(CeO<sub>2</sub>)<sub>10</sub> structures, obtained from a site scan for CH<sub>3</sub> adsorption, in relation to the most stable adsorption site of H specie. Figure 6. Relative reaction energy (Δ<i>E</i><sub>r</sub>) of (CH<sub><i>n</i></sub> + (4 – <i>n</i>)H)/(CeO<sub>2</sub>)<sub>10</sub> representative structures selected by <i>k</i>-means algorithm, with the aim to analyze the possibilities of the new compounds formation along dehydrogenation. Adsorption energy of CH<sub>3</sub> and H, <i>E</i><sub>ad</sub><sup>CH<sub>3</sub></sup> and <i>E</i><sub>ad</sub><sup>H</sup>, respectively; reaction enthalpy, Δ<i>H</i><sub>r</sub>; activation energy barrier, <i>E</i><sub>a</sub>; and relative reaction energy, Δ<i>E</i><sub>r</sub>. The ZPE corrections are represented by the value in parentheses. This article has not yet been cited by other publications.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"81 1","pages":""},"PeriodicalIF":3.3000,"publicationDate":"2024-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Correction to “Ab Initio Investigation of CH4 Dehydrogenation on a (CeO2)10 Cluster”\",\"authors\":\"Carina S. T. Peraça, Karla F. Andriani, Maurício J. Piotrowski, Juarez L. F. Da Silva\",\"doi\":\"10.1021/acs.jpcc.4c07452\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Unfortunately, we identified two errors in our paper related to the misapplication of the unity bond index–quadratic exponential potential (UBI–QEP) analysis, which became evident through discussions with Dr. Verónica Ganduglia-Pirovano and Dr. Breno L. Galvão (Private communication, March 15, 2024). The first error involves the results used to design Figure 5, where we obtained the activation energy values using the UBI-QEP approximation. In this analysis, we considered the most common adsorption sites reported in the literature for the CeO<sub>2</sub>(111) surface rather than the most stable ones on the ceria cluster. As a result, the dependence between the CH<sub>4</sub> adsorption site and the corresponding CH<sub>3</sub> and/or H sites was not maintained. A more appropriate approach for our purposes would be to conduct a systematic investigation of the coadsorption sites on (CeO<sub>2</sub>)<sub>10</sub>, using a specific adsorption site as a reference and mapping the most stable CH<sub>3</sub> or H sites. Therefore, we implemented the aforementioned approach by mapping the CH<sub>3</sub> sites and fixing the most stable H site as a reference; hence, the text and the related figure (Figure 5) on page 11944 should be updated based on the obtained new results. The second error concerns the formulation of Figure 6, where the transverse lines connecting the structures may give the reader the mistaken impression of a proposed reaction path. In reality, we present the reaction energy values for representative structures of each group selected by the <i>k-means</i> clustering method. Regarding the mistake in the production of Figures 5 and 6, the following changes must be applied: On page 11946, the sentence “The CH<sub>4</sub> molecule can adsorb on the nanocluster considering four adsorption configurations, [...]” must be replaced with “The CH<sub>4</sub> molecule can adsorb on the nanocluster considering three adsorption configurations, [...]” and the sentence “[...] C–H bond break on the (CeO<sub>2</sub>)<sub>10</sub> nanocluster can be related to the H adsorption site [...]” must be replaced with “[...] C–H bond break on the (CeO<sub>2</sub>)<sub>10</sub> nanocluster can be related to the coadsorption sites of CH<sub>3</sub> (H)[...]”. The original Figure 5 should be replaced by Figure 5 of this Correction. On page 11944, the sentence “We found four different adsorption modes for CH<sub>4</sub> on (CeO<sub>2</sub>)<sub>10</sub>, namely, umbrella, antiumbrella, scissoring, and modified antiumbrella, as indicated in Figure 5.<sup>66</sup> In the umbrella and antiumbrella modes, the calculated barrier (<i>E</i><sub>a</sub>) for the CH<sub>4</sub> dissociation (CH<sub>4</sub> → CH<sub>3</sub> + H*) indicates a higher cluster reactivity compared to the CeO<sub>2</sub>(111) surface. In this case, we obtained <i>E</i><sub>a</sub> equal to 0.54 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.48 eV) and 0.59 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.52 eV) for umbrella and antiumbrella modes, respectively, smaller than the value reported for the CeO<sub>2</sub>(111) surface (1.44 eV).<sup>65</sup>” must be replaced with “We found three different adsorption modes for CH<sub>4</sub> on (CeO<sub>2</sub>)<sub>10</sub> clusters, namely umbrella, antiumbrella, and scissoring.<sup>66</sup> The calculated barrier (<i>E</i><sub>a</sub>) for the CH<sub>4</sub> dissociation (CH<sub>4</sub> → CH<sub>3</sub> + H*) indicates a higher cluster reactivity compared to the CeO<sub>2</sub>(111) surface. In this case, we obtained <i>E</i><sub>a</sub> equal to 0.07 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.03) and 0.13 eV (<i>E</i><sub>a</sub><sup>ZPE</sup> = 0.09 eV) for the configuration proposed in Figure 1a,b, respectively, smaller than the value reported for the CeO<sub>2</sub>(111) surface (1.44 eV).<sup>65</sup>” In page 11944, the sentence “Then, the adsorption energy for the umbrella and antiumbrella modes in this state is −3.59 and −3.95 eV, respectively, in agreement with the value of −3.68 eV reported for CeO<sub>2</sub>(111) surface.<sup>25</sup>” must be removed. The original Figure 6 should be replaced by Figure 6 of this Correction. On page 11945, the following paragraph must be removed: “The energy barriers for the modified antiumbrella and scissoring modes (0.81 and 0.64 eV, respectively) indicate a higher barrier compared to the umbrella and antiumbrella modes, which can be associated with the H adsorption site. Once again, the ZPE corrections do not produce significant changes in the <i>E</i><sub>a</sub> values (0.74 and 0.56 eV, respectively). The difference between the modified and antiumbrella modes is the H adsorption site, since the former adsorbs in the 3-fold top<sup>O</sup> site, allowing the molecule adsorption at a 2-fold top<sup>O</sup> site. The opposite occurs for the latter one, in which the molecule adsorbs on the adsorption O site with a higher coordination. However, the activation energy obtained for the modified antiumbrella and scissoring mode can be compared with the values reported by Righi et al. for pristine CeO<sub>2</sub>(100) surface (0.87 eV).<sup>25</sup>” Table S48 has been corrected in the Supporting Information file and replaced by Table S48 here. The corrections mentioned do not affect the other discussions or our general conclusions. We regret any inconveniences. The authors thank Verónica Ganduglia–Pirovano and Breno L. Galvão for their valuable scientific discussions.<named-content content-type=\\\"anchor\\\" r type=\\\"simple\\\"></named-content><named-content content-type=\\\"anchor\\\" r type=\\\"simple\\\"></named-content><named-content content-type=\\\"anchor\\\" r type=\\\"simple\\\"></named-content> Figure 5. Relative reaction energy (Δ<i>E</i><sub>r</sub>) for the CH<sub>4</sub> → CH<sub>3</sub> + H step of CH<sub>4</sub> dehydrogenation. Frames (a) and (b) represent the two most stable (CH<sub>3</sub> + H)/(CeO<sub>2</sub>)<sub>10</sub> structures, obtained from a site scan for CH<sub>3</sub> adsorption, in relation to the most stable adsorption site of H specie. Figure 6. Relative reaction energy (Δ<i>E</i><sub>r</sub>) of (CH<sub><i>n</i></sub> + (4 – <i>n</i>)H)/(CeO<sub>2</sub>)<sub>10</sub> representative structures selected by <i>k</i>-means algorithm, with the aim to analyze the possibilities of the new compounds formation along dehydrogenation. Adsorption energy of CH<sub>3</sub> and H, <i>E</i><sub>ad</sub><sup>CH<sub>3</sub></sup> and <i>E</i><sub>ad</sub><sup>H</sup>, respectively; reaction enthalpy, Δ<i>H</i><sub>r</sub>; activation energy barrier, <i>E</i><sub>a</sub>; and relative reaction energy, Δ<i>E</i><sub>r</sub>. The ZPE corrections are represented by the value in parentheses. This article has not yet been cited by other publications.\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"81 1\",\"pages\":\"\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-11-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcc.4c07452\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c07452","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Correction to “Ab Initio Investigation of CH4 Dehydrogenation on a (CeO2)10 Cluster”
Unfortunately, we identified two errors in our paper related to the misapplication of the unity bond index–quadratic exponential potential (UBI–QEP) analysis, which became evident through discussions with Dr. Verónica Ganduglia-Pirovano and Dr. Breno L. Galvão (Private communication, March 15, 2024). The first error involves the results used to design Figure 5, where we obtained the activation energy values using the UBI-QEP approximation. In this analysis, we considered the most common adsorption sites reported in the literature for the CeO2(111) surface rather than the most stable ones on the ceria cluster. As a result, the dependence between the CH4 adsorption site and the corresponding CH3 and/or H sites was not maintained. A more appropriate approach for our purposes would be to conduct a systematic investigation of the coadsorption sites on (CeO2)10, using a specific adsorption site as a reference and mapping the most stable CH3 or H sites. Therefore, we implemented the aforementioned approach by mapping the CH3 sites and fixing the most stable H site as a reference; hence, the text and the related figure (Figure 5) on page 11944 should be updated based on the obtained new results. The second error concerns the formulation of Figure 6, where the transverse lines connecting the structures may give the reader the mistaken impression of a proposed reaction path. In reality, we present the reaction energy values for representative structures of each group selected by the k-means clustering method. Regarding the mistake in the production of Figures 5 and 6, the following changes must be applied: On page 11946, the sentence “The CH4 molecule can adsorb on the nanocluster considering four adsorption configurations, [...]” must be replaced with “The CH4 molecule can adsorb on the nanocluster considering three adsorption configurations, [...]” and the sentence “[...] C–H bond break on the (CeO2)10 nanocluster can be related to the H adsorption site [...]” must be replaced with “[...] C–H bond break on the (CeO2)10 nanocluster can be related to the coadsorption sites of CH3 (H)[...]”. The original Figure 5 should be replaced by Figure 5 of this Correction. On page 11944, the sentence “We found four different adsorption modes for CH4 on (CeO2)10, namely, umbrella, antiumbrella, scissoring, and modified antiumbrella, as indicated in Figure 5.66 In the umbrella and antiumbrella modes, the calculated barrier (Ea) for the CH4 dissociation (CH4 → CH3 + H*) indicates a higher cluster reactivity compared to the CeO2(111) surface. In this case, we obtained Ea equal to 0.54 eV (EaZPE = 0.48 eV) and 0.59 eV (EaZPE = 0.52 eV) for umbrella and antiumbrella modes, respectively, smaller than the value reported for the CeO2(111) surface (1.44 eV).65” must be replaced with “We found three different adsorption modes for CH4 on (CeO2)10 clusters, namely umbrella, antiumbrella, and scissoring.66 The calculated barrier (Ea) for the CH4 dissociation (CH4 → CH3 + H*) indicates a higher cluster reactivity compared to the CeO2(111) surface. In this case, we obtained Ea equal to 0.07 eV (EaZPE = 0.03) and 0.13 eV (EaZPE = 0.09 eV) for the configuration proposed in Figure 1a,b, respectively, smaller than the value reported for the CeO2(111) surface (1.44 eV).65” In page 11944, the sentence “Then, the adsorption energy for the umbrella and antiumbrella modes in this state is −3.59 and −3.95 eV, respectively, in agreement with the value of −3.68 eV reported for CeO2(111) surface.25” must be removed. The original Figure 6 should be replaced by Figure 6 of this Correction. On page 11945, the following paragraph must be removed: “The energy barriers for the modified antiumbrella and scissoring modes (0.81 and 0.64 eV, respectively) indicate a higher barrier compared to the umbrella and antiumbrella modes, which can be associated with the H adsorption site. Once again, the ZPE corrections do not produce significant changes in the Ea values (0.74 and 0.56 eV, respectively). The difference between the modified and antiumbrella modes is the H adsorption site, since the former adsorbs in the 3-fold topO site, allowing the molecule adsorption at a 2-fold topO site. The opposite occurs for the latter one, in which the molecule adsorbs on the adsorption O site with a higher coordination. However, the activation energy obtained for the modified antiumbrella and scissoring mode can be compared with the values reported by Righi et al. for pristine CeO2(100) surface (0.87 eV).25” Table S48 has been corrected in the Supporting Information file and replaced by Table S48 here. The corrections mentioned do not affect the other discussions or our general conclusions. We regret any inconveniences. The authors thank Verónica Ganduglia–Pirovano and Breno L. Galvão for their valuable scientific discussions. Figure 5. Relative reaction energy (ΔEr) for the CH4 → CH3 + H step of CH4 dehydrogenation. Frames (a) and (b) represent the two most stable (CH3 + H)/(CeO2)10 structures, obtained from a site scan for CH3 adsorption, in relation to the most stable adsorption site of H specie. Figure 6. Relative reaction energy (ΔEr) of (CHn + (4 – n)H)/(CeO2)10 representative structures selected by k-means algorithm, with the aim to analyze the possibilities of the new compounds formation along dehydrogenation. Adsorption energy of CH3 and H, EadCH3 and EadH, respectively; reaction enthalpy, ΔHr; activation energy barrier, Ea; and relative reaction energy, ΔEr. The ZPE corrections are represented by the value in parentheses. This article has not yet been cited by other publications.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


