通过第一原理元动力学模拟阐明 PEO/(101)-TiO2 Anatase界面的结构和电子特性
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
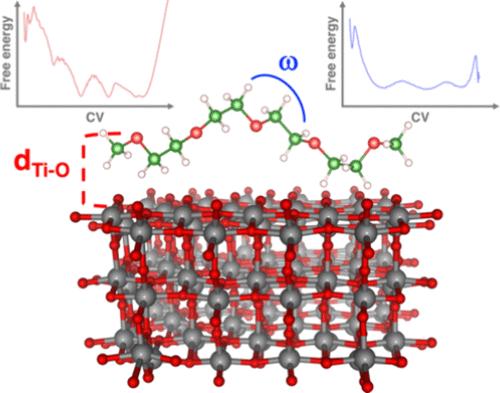
通过元动力学模拟和密度泛函紧密结合,探索了聚环氧乙烷/金刚石界面(即 PEO/TiO2)的结构和动态特性。1-ns 轨迹是根据两个不同的结构相关集合变量重建的:界面 Tisurf-OPEO 距离和聚合物链扭转角。PEO 的构象自由度受到与锐钛矿表面不饱和钛位点的多种有利相互作用的显著影响。根据这些轨迹,利用密度泛函理论中的电子结构计算分析了从自由能表面提取的几种平衡结构:钛白粉的功函数结果在很大程度上受到锐钛矿表面上 PEO 动态解析结构的影响。除了对与技术相关的 PEO/TiO2 界面有内在的宝贵见解外,我们的研究结果还提供了第一个创新实例,用经济可靠的计算协议来描述异质界面并预测分子动力学对相关理化性质的影响。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Elucidating Structural and Electronic Features of PEO/(101)-TiO2 Anatase Interfaces through First-Principles Metadynamics Simulations
Structural and dynamic properties of the poly(ethylene oxide)/anatase interface (i.e., PEO/TiO2) are explored via metadynamics simulations and density functional tight binding. 1-ns trajectories are reconstructed upon two different structure-related collective variables: interfacial Tisurf–OPEO distances and polymer chain torsion angles. The conformational freedom of PEO is significantly influenced by multiple favorable interactions with unsaturated Ti sites on the anatase surface. From these trajectories, several equilibrium structures extracted from the free-energy surface are analyzed using electronic structure calculations within density functional theory: the titania work function results in being largely influenced by the dynamically resolved structuring of PEO on the anatase surface. Besides the intrinsic valuable insights into the technologically relevant PEO/TiO2 interface, our findings provide the first innovative example of an affordable yet reliable computational protocol to describe a heterogeneous interface and predict the effects of molecular dynamics on relevant physicochemical properties.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


