设计和研究卟啉席夫碱纳米片的电子状态,K+ - 电池的阴极材料
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
由于电池技术缺乏有效的高通量筛选方法,为钾离子电池(KIB)确定新电极材料仍然十分困难。KIB 的耐用性、经济性、安全性以及与锂离子电池的相似性使其获得了极大的关注。卟啉基材料因其较大的表面积和有利的光物理特性而成为具有吸引力的选择。作为 KIB 的有效阴极材料,本研究提出了卟啉希夫碱纳米结构 (SBN)。我们的目标是利用密度泛函理论(DFT)和高斯 09 程序以及 B3LYP/6-31G(d) 基集,改进掺杂钾离子的卟啉衍生物。我们还计算了重要的电子参数,如带隙、亲电性、化学势和前沿轨道(HOMO 和 LUMO)。经测定,化合物 1 至 4 的 HOMO-LUMO 间隙分别为 2.97、1.52、1.38 和 1.36。计算得出的间隙表明,化合物的反应性从 1 到 4 越来越高,而稳定性则从 1 到 4 越低。基于这些计算观察结果,预计本理论分析将为未来研究人员通过实验探索这些化合物的实际应用提供依据。本文章由计算机程序翻译,如有差异,请以英文原文为准。
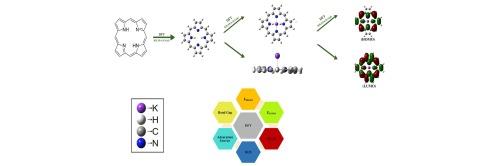
Designing and investigating electronic states of porphyrin Schiff bases nanoflakes, cathode materials for K+ - batteries
Identifying new electrode materials for K-ion batteries (KIBs) is still difficult since battery technology lacks an effective high-throughput screening approach. The durability, affordability, safety, and resemblance to Li-ion batteries of KIBs have garnered them tremendous attention. Porphyrin-based materials have become attractive options because of their generous surface area and advantageous photo-physical characteristics. As effective cathodic materials for KIBs, porphyrin Schiff base nanostructures (SBNs) are suggested in this work. Our goal was to improve Potassium-ion doped porphyrin derivatives by using Density Functional Theory (DFT) with the Gaussian 09 program with a B3LYP/6-31G(d) basis set. We also calculated important electronic parameters such as the band gap, electrophilicity, chemical potential, and frontier orbitals (HOMO and LUMO). The determined HOMO-LUMO gaps for compounds 1 to 4 were 2.97, 1.52, 1.38, and 1.36 respectively. The computed gaps indicate that the reactivity of the compounds increases from 1 to 4, whereas the stability decreases from 1 to 4.
Based on these computational observations, it is expected that this theoretical analysis will provide a basis for future researchers to explore the practical applications of these compounds through experimentation.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


