通过量子计算机计算分子电子结构
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
量子计算机可用于计算多电子分子系统的电子结构和估计基态能量。在本研究中,我们采用了变分量子均衡器(VQE)算法,作为一种量子-经典混合算法来计算量子比特数呈上升趋势的 H3+、OH-、HF 和 BH3 等分子的基态能。我们利用费米子到量子比特编码的奇偶校验变换和单双激发单元耦合簇(UCCSD)来构建一个等式。我们将量子模拟结果与计算化学方法进行了比较,包括作为基准能量的全构型相互作用(FCI)和作为常用计算方法的非限制哈特里-福克(UHF)。我们的结果表明,VQE 和 FCI 得出的分子基态能量具有良好的一致性。此外,在我们的工作中,通过 VQE 获得的基态能的精确度高于之前报道的值。这项工作的目的是对 VQE 算法进行基准测试,以计算一组新分子的电子基态能,这些分子可以很好地在实际量子计算机上进行分子模拟。本文章由计算机程序翻译,如有差异,请以英文原文为准。
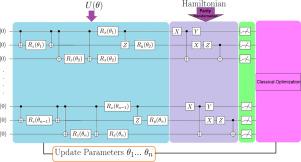
Molecular electronic structure calculation via a quantum computer
Quantum computers can be used to calculate the electronic structure and estimate the ground state energy of many-electron molecular systems. In the present study, we implement the Variational Quantum Eigensolver (VQE) algorithm, as a hybrid quantum–classical algorithm to calculate the ground state energy of the molecules such as H, OH, HF and BH in which the number of qubits has an increasing trend. We use the parity transformation for fermion to qubit encoding and the Unitary Coupled Cluster for Single and Double excitations (UCCSD) to construct an ansatz. We compare our quantum simulation results with the computational chemistry approaches including Full Configuration Interaction (FCI), as benchmark energy and Unrestricted Hartree–Fock (UHF), as a common computational method. Our results show that there is a good agreement between molecular ground state energy obtained from VQE and FCI. Moreover, the accuracy of the ground state energies obtained from VQE in our work is higher than the previously reported values. This work aims to benchmark the VQE algorithm to calculate the electronic ground state energy for a new set of molecules that can be good candidates for molecular simulation on a real quantum computer.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


