基于状态密度的描述符,用于晶界偏析能的高效机器学习
IF 3.1
3区 材料科学
Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
摘要
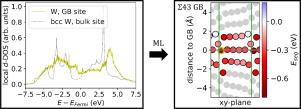
合金元素在晶界(GB)的偏析对材料的机械和功能特性有重大影响。这一过程受偏析能的控制,而偏析能可通过非线性方法精确计算。在过去几年中,为了降低计算成本,模拟计算已经与机器学习(ML)方法相结合。在这里,我们展示了如何将电子结构信息纳入 ML。为了获得电子结构,我们使用了两种方法:(i) 密度泛函理论(DFT);(ii) 紧结合(TB)哈密顿的递归解。利用推导出的描述符,我们对来自 15 个重合位点晶格 GB(Σ 值最高可达 43)的非原位偏析数据训练了一个线性模型和一个高斯过程,并利用交叉验证得分对模型进行了比较。结果发现,TB 和 DFT 衍生的描述符都明显优于以前用于 ML 离析能的普通结构特征。此外,TB 描述符几乎达到了与 DFT 描述符相同的精度,但其计算量却大大减少。我们在 bcc-W 矩阵中将 Ta 和 Re 分离成 GBs 的过程中测试了我们的方法,这些材料与聚变能研究息息相关。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Descriptors based on the density of states for efficient machine learning of grain-boundary segregation energies
The segregation of alloying elements to grain-boundaries (GB) has a significant impact on mechanical and functional properties of materials. The process is controlled by the segregation energies, that can accurately be computed using ab-initio methods. Over the last years, ab-initio computations have been combined with machine-learning (ML) approaches for a reduction of computational cost. Here, we show how information from the electronic structure can be incorporated in the ML. To obtain the electronic structure we use two methods, (i) density functional theory (DFT), and (ii) a recursive solution of a tight-binding (TB) Hamiltonian. With the derived descriptors we train a linear model and a Gaussian process on ab-initio segregation data from 15 coincident site lattice GBs with -values up to 43, where the models are compared using cross-validation scores. Both the TB and DFT-derived descriptors are found to clearly outperform common structure-based features that have been used for ML segregation energies before. Furthermore, TB descriptors almost reach the same accuracy as DFT descriptors although their computational effort is significantly reduced. We test our approach on segregation of Ta and Re to GBs in a bcc-W matrix, which are materials of relevance for fusion-energy research.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Materials Science
工程技术-材料科学:综合
CiteScore
6.50
自引率
6.10%
发文量
665
审稿时长
26 days
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


