潜在抗病毒药物的计算研究和抗菌活性预测
IF 4
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
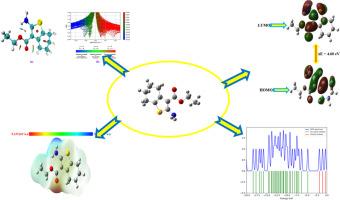
由于 2-氨基-4,5,6,7-四氢苯并[b]噻吩-3-羧酸乙酯(S1)具有重要的生物和药理活性,因此被选作本次研究的对象。我们从理论和实验两方面对其进行了详细的红外光谱和拉曼光谱研究。对 S1 的前沿分子轨道和电子特性进行了解释。在分子静电位(MEP)图中,NO2 基团呈现蓝色,表明存在亲核位点。此外,还研究了状态密度(DOS)、非共价相互作用(NCI)、电子局部功能(ELF)、局部轨道定位器(LOL)、Mulliken 原子电荷和反应位点。还对抗菌、抗真菌、抗毒素、抗病毒和抗霉菌进行了研究。此外,还对该化合物进行了分子对接研究。利用 Auto-dock 程序对化合物 S1 进行了分子对接研究。分子对接研究中,4XGK、4ATO、3U9 G 和 2DP4 的最低结合能分别为 -7.91、-5.62、-6.92 和 -5.85 kcal/mol。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Computational investigation and antimicrobial activity prediction of potential antiviral drug
The ethyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (S1) was chosen for this investigation due to its important biological and pharmacological activities. Detailed infrared and Raman spectra have been investigated in both theoretical and experimental. Frontier molecular orbitals and the electronic properties of the S1 are explained. The titled compound S1 calculated HOMO-LUMO energy gap is 4.60 eV In molecular electrostatic potential (MEP) map the NO2 group exhibits a blue color, indicating the presence of nucleophilic sites. The density of state (DOS), non-covalent interaction (NCI), electron localized function (ELF), localized orbital locator (LOL), Mulliken atomic charges, and reactive sites have been investigated. The anti-bacterial, antifungal, antitoxin, antiviral, and antimycobacterial studies have been investigated. The molecular docking investigations of the compound were also conducted. Using the Auto-dock program, the compound S1 molecular docking study has been conducted. The molecular docking study's lowest binding energy is -7.91, -5.62, -6.92, and -5.85 kcal/mol for 4XGK, 4ATO, 3U9 G, and 2DP4 respectively.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Molecular Structure
化学-物理化学
CiteScore
7.10
自引率
15.80%
发文量
2384
审稿时长
45 days
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


