HiMolformer:整合图形和序列表示法,利用 SMILES 预测肝脏微粒体的稳定性。
IF 2.6
4区 生物学
Q2 BIOLOGY
引用次数: 0
摘要
在药物发现或临床前研究的初始阶段,了解新分子的代谢稳定性至关重要。最近,用于分子性质预测的预训练深度学习研究取得了积极进展,各种模型已被开源。然而,这些模型大多依赖于二维图或一维序列进行训练,而且所使用的数据格式不同,表示方法也不尽相同。因此,结合多种表示方法可以拓宽学习范围,并有可能成为一种易于管理且最有效的提高性能的方法。因此,我们提出了一种预测代谢稳定性的新型混合模型,该模型综合了基于图和基于序列的模型的表征,并针对分子特征进行了预先训练。这种方法综合利用了分子的二维拓扑信息和一维序列信息。我们选择了基于图的图神经网络(GNN)模型 HiMol 和基于序列的 Transformer 模型 Molformer 进行整合,因此将其命名为 HiMolformer。与其他模型相比,HiMolformer 表现出了卓越的性能。我们还重点利用韩国化学库(KCB)的经验数据集进行回归预测,该数据集包括从实际代谢反应实验中获得的小鼠肝脏微粒体(MLM)和人类肝脏微粒体(HLM)数据,共 3498 个分子。据我们所知,这是首次尝试利用单一 SMILES 输入使用回归法开发 MLM 和 HLM 预测模型。该模型的源代码见 https://github.com/YUNSEOKWOO/HiMolformer。本文章由计算机程序翻译,如有差异,请以英文原文为准。
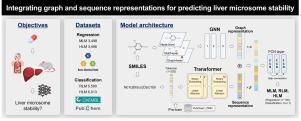
HiMolformer: Integrating graph and sequence representations for predicting liver microsome stability with SMILES
In the initial stages of drug discovery or pre-clinical studies, understanding the metabolic stability of new molecules is crucial. Recently, research on pre-trained deep learning for molecular property prediction has been actively progressing, with various models being made open-source. However, most of these models rely on either 2D graph or 1D sequence for training, and the representation varies depending on the data format used. Consequently, combining multiple representations can broaden the scope of learning and may potentially be a manageable and most effective method to enhance performance.
Therefore, we propose a novel hybrid model for predicting metabolic stability, which integrates representations from both graph-based and sequence-based models pre-trained for molecular features. This approach utilizes the combined strengths of 2D topological and 1D sequential information of molecules. HiMol, a graph-based graph neural network (GNN) model, and Molformer, a sequence-based Transformer model, were selected for integration, thus we named it HiMolformer. HiMolformer demonstrated superior performance compared to other models. We also focus on regression task for prediction with a empirical dataset from Korea Chemical Bank (KCB), comprising 3,498 molecules with mouse liver microsome (MLM) and human liver microsome (HLM) data obtained from actual metabolic reaction experiments. To the best of our knowledge, it is the first attempt to develop MLM and HLM prediction models using regression with a single SMILES input. The source code of this model is available at https://github.com/YUNSEOKWOO/HiMolformer.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


