通过异噁唑啉环加载物的电化学活化实现烯烃 1,2-氰基-羟基化的一般步骤
IF 14.4
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
立体选择性烯烃 1,2-二官能化是一种从现成的 "扁平 "碳原料中获得富含三维 C(sp3)手性分子的独特策略。最先进的方法是利用手性过渡金属催化剂实现高度的区域和立体控制。然而,这往往是以烯烃范围有限和通用性降低为代价的。1,3-二极环化反应通常用于从烯形成杂环,具有高度的区域选择性和立体特异性。然而,环加成产物的开环方法需要使用危险试剂或强制反应条件,从而限制了其合成应用。在本文中,我们介绍了一种实用、通用和选择性的烯烃 1,2-同步双官能化电合成策略的实施情况,该策略取决于新型试剂的设计--该试剂由带有磺酰基手柄的 1,3-二极氧化腈前体组成。这些试剂可以通过 "点击 "1,3-二极环化反应选择性地使烯烃发生双官能化,然后促进随后产生的异噁唑啉中间体的伸缩电化学单电子转移活化。环加成物的阴极还原引发自由基碎片途径,从而产生所需的立体定向 1,2-syn-hydroxy 腈衍生物。我们的伸缩电化学过程可容忍广泛的官能度,而且能使富电子、贫电子和未活化的烯烃以及具有不同取代度的烯烃发生二官能化反应,从而为当前的烯烃二官能化策略提供了一种稳健、通用和选择性强的无金属替代方法。利用这些特点,我们采用电合成方法实现了天然产物和生物活性化合物的后期合成-羟基-氰化,并简化了药物的从头合成。本文章由计算机程序翻译,如有差异,请以英文原文为准。
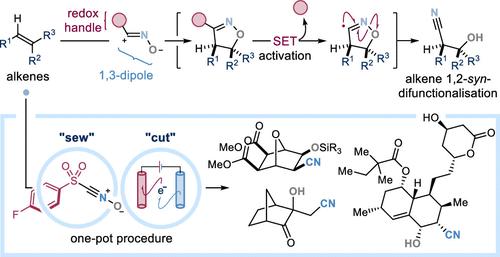
General Alkene 1,2-syn-Cyano-Hydroxylation Procedure Via Electrochemical Activation of Isoxazoline Cycloadducts
Stereoselective alkene 1,2-difunctionalization is a privileged strategy to access three-dimensional C(sp3)-rich chiral molecules from readily available “flat” carbon feedstocks. State-of-the-art approaches exploit chiral transition metal-catalysts to enable high levels of regio- and stereocontrol. However, this is often achieved at the expense of a limited alkene scope and reduced generality. 1,3-Dipolar cycloadditions are routinely used to form heterocycles from alkenes with high levels of regioselectivity and stereospecificity. Nevertheless, methods for the ring-opening of cycloadducts to reveal synthetically useful functionalities require the use of hazardous reagents or forcing reaction conditions; thus limiting their synthetic applications. Herein, we describe the implementation of a practical, general and selective electrosynthetic strategy for olefin 1,2-syn-difunctionalization, which hinges on the design of novel reagents–consisting of a nitrile oxide 1,3-dipole precursor, equipped with a sulfonyl-handle. These can selectively difunctionalize alkenes via “click” 1,3-dipolar cycloadditions, and then facilitate the telescoped electrochemical single electron transfer activation of the ensuing isoxazoline intermediate. Cathodic reduction of the cycloadduct triggers a radical fragmentation pathway delivering sought-after stereodefined 1,2-syn-hydroxy nitrile derivatives. Our telescoped electrochemical procedure tolerates a wide range of functionalities, and─crucially─enables the difunctionalization of both electron-rich, electron-poor and unactivated olefins, with diverse degree of substitution; thus providing a robust, general and selective metal-free alternative to current alkene difunctionalization strategies. Capitalizing on these features, we employed our electrosynthetic method to enable the late-stage syn-hydroxy-cyanation of natural products and bioactive compounds, and streamline the de novo synthesis of pharmaceutical agents.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


