Ankit Shah, Huiting Xu, Hyok Joon Kwon, Fredric E. Wondisford
{"title":"甘油激酶缺乏症患者体内的甘油代谢。","authors":"Ankit Shah, Huiting Xu, Hyok Joon Kwon, Fredric E. Wondisford","doi":"10.1002/jmd2.12452","DOIUrl":null,"url":null,"abstract":"<p>Glycerol kinase deficiency (GKD) is an X-linked recessive disorder due to <i>glycerol kinase</i> (<i>GK</i>) gene mutations resulting in hyperglycerolermia, hyperglyceroluria, and “pseudohypertriglyceridemia.” In vivo glycerol metabolism has not been assessed in GKD. A 62-year-old man with suspected GKD and his extended family underwent whole exome sequencing and fasting blood work with two modes of lipid measurements: (1) standard lipase-based methodology and (2) nuclear magnetic resonance (NMR). Two overnight fasted men with GKD and a heterozygote female carrier then underwent <sup>13</sup>C<sub>3</sub>-glycerol infusion. Affected family members had a novel two-nucleotide deletion in exon 5 of the <i>GK</i> gene (c.373_374del). Compared to their family members (<i>n</i> = 14), men with GKD (<i>n</i> = 5) had significantly lower total cholesterol levels (3.72 ± 0.70 vs. 4.77 ± 0.85 mmol/L, <i>p</i> = 0.024). Compared to NMR, lipase-based assays overreported triglycerides (5.28 ± 1.38 vs. 0.81 ± 0.32, mmol/L, <i>p</i> < 0.001) and underreported low-density lipoprotein cholesterol values (0.93 ± 0.23 vs. 2.18 ± 0.42 mmol/L, <i>p</i> = 0.001) in GKD. Men with GKD could not convert glycerol into glucose or triglycerides, which was preserved in the heterozygote carrier. Glycolytic metabolism of glycerol to lactate persisted in GKD, but it was reduced by a magnitude and, possibly, due to homologous glycerol kinases encoded by other genes. In summary, we report a novel <i>GK</i> pathogenic variant; affected men cannot convert circulating glycerol to glucose or triglycerides and have lower cholesterol levels. These results offer a human model for potentially targeting glycerol kinase to treat conditions associated with hyperglycemia and hyperlipidemia and warrant further investigation.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 6","pages":"392-400"},"PeriodicalIF":1.8000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11540572/pdf/","citationCount":"0","resultStr":"{\"title\":\"In vivo glycerol metabolism in patients with glycerol kinase deficiency\",\"authors\":\"Ankit Shah, Huiting Xu, Hyok Joon Kwon, Fredric E. Wondisford\",\"doi\":\"10.1002/jmd2.12452\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Glycerol kinase deficiency (GKD) is an X-linked recessive disorder due to <i>glycerol kinase</i> (<i>GK</i>) gene mutations resulting in hyperglycerolermia, hyperglyceroluria, and “pseudohypertriglyceridemia.” In vivo glycerol metabolism has not been assessed in GKD. A 62-year-old man with suspected GKD and his extended family underwent whole exome sequencing and fasting blood work with two modes of lipid measurements: (1) standard lipase-based methodology and (2) nuclear magnetic resonance (NMR). Two overnight fasted men with GKD and a heterozygote female carrier then underwent <sup>13</sup>C<sub>3</sub>-glycerol infusion. Affected family members had a novel two-nucleotide deletion in exon 5 of the <i>GK</i> gene (c.373_374del). Compared to their family members (<i>n</i> = 14), men with GKD (<i>n</i> = 5) had significantly lower total cholesterol levels (3.72 ± 0.70 vs. 4.77 ± 0.85 mmol/L, <i>p</i> = 0.024). Compared to NMR, lipase-based assays overreported triglycerides (5.28 ± 1.38 vs. 0.81 ± 0.32, mmol/L, <i>p</i> < 0.001) and underreported low-density lipoprotein cholesterol values (0.93 ± 0.23 vs. 2.18 ± 0.42 mmol/L, <i>p</i> = 0.001) in GKD. Men with GKD could not convert glycerol into glucose or triglycerides, which was preserved in the heterozygote carrier. Glycolytic metabolism of glycerol to lactate persisted in GKD, but it was reduced by a magnitude and, possibly, due to homologous glycerol kinases encoded by other genes. In summary, we report a novel <i>GK</i> pathogenic variant; affected men cannot convert circulating glycerol to glucose or triglycerides and have lower cholesterol levels. These results offer a human model for potentially targeting glycerol kinase to treat conditions associated with hyperglycemia and hyperlipidemia and warrant further investigation.</p>\",\"PeriodicalId\":14930,\"journal\":{\"name\":\"JIMD reports\",\"volume\":\"65 6\",\"pages\":\"392-400\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-09-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11540572/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JIMD reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12452\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12452","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
In vivo glycerol metabolism in patients with glycerol kinase deficiency
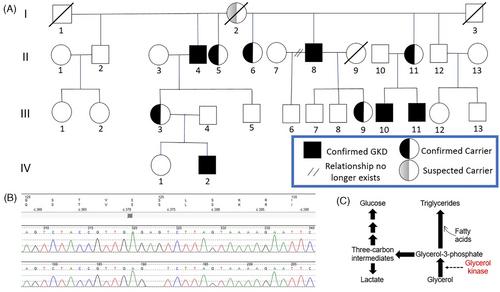
Glycerol kinase deficiency (GKD) is an X-linked recessive disorder due to glycerol kinase (GK) gene mutations resulting in hyperglycerolermia, hyperglyceroluria, and “pseudohypertriglyceridemia.” In vivo glycerol metabolism has not been assessed in GKD. A 62-year-old man with suspected GKD and his extended family underwent whole exome sequencing and fasting blood work with two modes of lipid measurements: (1) standard lipase-based methodology and (2) nuclear magnetic resonance (NMR). Two overnight fasted men with GKD and a heterozygote female carrier then underwent 13C3-glycerol infusion. Affected family members had a novel two-nucleotide deletion in exon 5 of the GK gene (c.373_374del). Compared to their family members (n = 14), men with GKD (n = 5) had significantly lower total cholesterol levels (3.72 ± 0.70 vs. 4.77 ± 0.85 mmol/L, p = 0.024). Compared to NMR, lipase-based assays overreported triglycerides (5.28 ± 1.38 vs. 0.81 ± 0.32, mmol/L, p < 0.001) and underreported low-density lipoprotein cholesterol values (0.93 ± 0.23 vs. 2.18 ± 0.42 mmol/L, p = 0.001) in GKD. Men with GKD could not convert glycerol into glucose or triglycerides, which was preserved in the heterozygote carrier. Glycolytic metabolism of glycerol to lactate persisted in GKD, but it was reduced by a magnitude and, possibly, due to homologous glycerol kinases encoded by other genes. In summary, we report a novel GK pathogenic variant; affected men cannot convert circulating glycerol to glucose or triglycerides and have lower cholesterol levels. These results offer a human model for potentially targeting glycerol kinase to treat conditions associated with hyperglycemia and hyperlipidemia and warrant further investigation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


