{"title":"无症状的儿科 S-腺苷高半胱氨酸水解酶缺乏症。","authors":"Patrícia Lipari Pinto, Marjorie Dixon, Sniya Sudhakar, Ivo Baric, Julien Baruteau","doi":"10.1002/jmd2.12449","DOIUrl":null,"url":null,"abstract":"<p>S-adenosylhomocysteine hydrolase deficiency is an autosomal recessive inborn error of metabolism affecting methylation by disrupting the methionine cycle. Its clinical spectrum spans from severe perinatal encephalomyopathy and liver failure to asymptomatic course in patients with isolated hypermethioninemia. We present two new cases of S-adenosylhomocysteine hydrolase deficiency from Pakistani origin clinically asymptomatic at presentation. Both siblings showed mild chronic liver failure and elevation of creatine kinase. The older patient presented at 6 years of age with isolated verbal processing difficulty and mild diffuse leukodystrophy, reversible 12 months after introduction of methionine dietary restriction. The patient showed subtle atrophy in the muscle MRI at the age of 7 years. S-adenosylhomocysteine hydrolase deficiency was confirmed with homozygous missense variant c.146G>A (p.Arg49His) in the <i>AHCY</i> gene, a genotype previously reported in Pakistani patients with mild presentation. Dietary methionine restriction decreased plasma methionine but not plasma S-adenosylhomocysteine and S-adenosylmethionine. This work expands the mild spectrum of S-adenosylhomocysteine hydrolase deficiency with no noticeable clinical symptoms in children, highlighting a specific hotspot variant from South Asia. This mild form of the disease is likely underdiagnosed and raises the question of therapeutic management to prevent long-term complications documented in the literature, such as hepatocellular carcinoma and myopathy in early adulthood.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 6","pages":"371-381"},"PeriodicalIF":1.8000,"publicationDate":"2024-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11540567/pdf/","citationCount":"0","resultStr":"{\"title\":\"Asymptomatic pediatric presentation of S-adenosylhomocysteine hydrolase deficiency\",\"authors\":\"Patrícia Lipari Pinto, Marjorie Dixon, Sniya Sudhakar, Ivo Baric, Julien Baruteau\",\"doi\":\"10.1002/jmd2.12449\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>S-adenosylhomocysteine hydrolase deficiency is an autosomal recessive inborn error of metabolism affecting methylation by disrupting the methionine cycle. Its clinical spectrum spans from severe perinatal encephalomyopathy and liver failure to asymptomatic course in patients with isolated hypermethioninemia. We present two new cases of S-adenosylhomocysteine hydrolase deficiency from Pakistani origin clinically asymptomatic at presentation. Both siblings showed mild chronic liver failure and elevation of creatine kinase. The older patient presented at 6 years of age with isolated verbal processing difficulty and mild diffuse leukodystrophy, reversible 12 months after introduction of methionine dietary restriction. The patient showed subtle atrophy in the muscle MRI at the age of 7 years. S-adenosylhomocysteine hydrolase deficiency was confirmed with homozygous missense variant c.146G>A (p.Arg49His) in the <i>AHCY</i> gene, a genotype previously reported in Pakistani patients with mild presentation. Dietary methionine restriction decreased plasma methionine but not plasma S-adenosylhomocysteine and S-adenosylmethionine. This work expands the mild spectrum of S-adenosylhomocysteine hydrolase deficiency with no noticeable clinical symptoms in children, highlighting a specific hotspot variant from South Asia. This mild form of the disease is likely underdiagnosed and raises the question of therapeutic management to prevent long-term complications documented in the literature, such as hepatocellular carcinoma and myopathy in early adulthood.</p>\",\"PeriodicalId\":14930,\"journal\":{\"name\":\"JIMD reports\",\"volume\":\"65 6\",\"pages\":\"371-381\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11540567/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JIMD reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12449\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12449","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Asymptomatic pediatric presentation of S-adenosylhomocysteine hydrolase deficiency
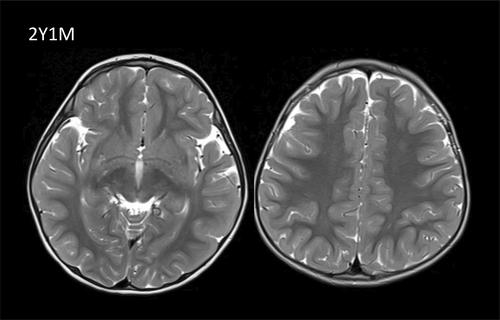
S-adenosylhomocysteine hydrolase deficiency is an autosomal recessive inborn error of metabolism affecting methylation by disrupting the methionine cycle. Its clinical spectrum spans from severe perinatal encephalomyopathy and liver failure to asymptomatic course in patients with isolated hypermethioninemia. We present two new cases of S-adenosylhomocysteine hydrolase deficiency from Pakistani origin clinically asymptomatic at presentation. Both siblings showed mild chronic liver failure and elevation of creatine kinase. The older patient presented at 6 years of age with isolated verbal processing difficulty and mild diffuse leukodystrophy, reversible 12 months after introduction of methionine dietary restriction. The patient showed subtle atrophy in the muscle MRI at the age of 7 years. S-adenosylhomocysteine hydrolase deficiency was confirmed with homozygous missense variant c.146G>A (p.Arg49His) in the AHCY gene, a genotype previously reported in Pakistani patients with mild presentation. Dietary methionine restriction decreased plasma methionine but not plasma S-adenosylhomocysteine and S-adenosylmethionine. This work expands the mild spectrum of S-adenosylhomocysteine hydrolase deficiency with no noticeable clinical symptoms in children, highlighting a specific hotspot variant from South Asia. This mild form of the disease is likely underdiagnosed and raises the question of therapeutic management to prevent long-term complications documented in the literature, such as hepatocellular carcinoma and myopathy in early adulthood.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


