用于预测中性物种溶解自由能的各种分子特征表示的比较研究。
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
在神经网络的帮助下预测分子特性是替代或增强综合量子化学计算的常用方法。研究人员面临的问题之一是如何选择矢量化方法来表示估算模型中的溶剂和溶质。在这项工作中,针对有机溶质和溶剂研究了 10 种不同的方法,包括依赖宏观参数、官能团分类、分子图或原子坐标的矢量化方法。在 MNSol 数据库上训练的名为 "JustBonds "的 "Bag of Bonds "方法变体显示出最佳的整体性能,其 RMSD本文章由计算机程序翻译,如有差异,请以英文原文为准。
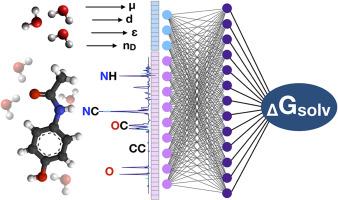
Comparative study of various molecular feature representations for solvation free energy predictions of neutral species
Predicting molecular properties with the help of Neural Networks is a common way to substitute or enhance comprehensive quantum-chemical calculations. One of the problems facing researchers is the choice of vectorization approach to representing the solvent and the solute for the estimator model. In this work, 10 different approaches have been investigated for both organic solute and solvent including vectorizers that relied on macroscopic parameters, functional groups classification, molecular graphs, or atomic coordinates. A variation of the Bag of Bonds approach called JustBonds, trained on the MNSol database, showed the best overall performance resulting in RMSD <2 kcal/mol for the blind dataset that contains the solutes not presented in the training subset and <1 kcal/mol on records from Solv@TUM database, which is close to contemporary continuum models. We have also demonstrated that the most important bags usually contain heteroatom and play a key role in the solvation process. Furthermore, the small role of solvent vectorization was demonstrated and revealed that approaches based on functional groups or macroscopic solvent parameters are often enough to efficiently represent solvent media.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


