{"title":"短读数测序数据中的串联重复序列分析:从已知致病性重复序列的基因分型到发现新的扩展。","authors":"Andreas Halman, Andrew Lonsdale, Alicia Oshlack","doi":"10.1002/cpz1.70010","DOIUrl":null,"url":null,"abstract":"<p>Short tandem repeats (STRs) and variable-number tandem repeats (VNTRs) are repetitive genomic sequences seen widely throughout the genome. These repeat expansions are currently known to cause ∼60 diseases, with expansions in new loci linked to rare diseases continuing to be discovered. Genome sequencing is an important tool for detecting disease-causing variants and several computational tools have been developed to analyze tandem repeats from genomic data, enabling the genotyping and the identification of expanded alleles. However, guidelines for conducting the analysis of these repeats and, more importantly, for assessing the findings are lacking. Understanding the tools and their technical limitations is important for accurately interpreting the results. This article provides detailed, step-by-step instructions for three key use cases in STR analysis from short-read genome sequencing data, which are also applicable to smaller VNTRs. First, it demonstrates an approach for genotyping known pathogenic loci and the identification of clinically significant expansions. Second, we offer guidance on defining tandem repeat loci and conducting genome-wide genotyping studies, which is also applicable to diploid organisms other than humans. Third, instructions are provided on how to find novel expansions at loci not previously known to be associated with disease, aiding in the discovery of new pathogenic loci. Moreover, we introduce the use of newly-developed helper tools that enable a complete and streamlined tandem repeat analysis protocol by addressing the gaps in current methods. All three protocols are compatible with human hg19, hg38, and the latest telomere-to-telomere (hs1) reference genomes. Additionally, this protocol provides an overview and discussion on how to interpret genotyping results. © 2024 The Author(s). Current Protocols published by Wiley Periodicals LLC.</p><p><b>Basic Protocol 1</b>: Genotyping known pathogenic tandem repeat loci</p><p><b>Alternate Protocol</b>: Genotyping known pathogenic tandem repeat loci with STRipy</p><p><b>Support Protocol 1</b>: Installation of tools and ExpansionHunter catalog modification</p><p><b>Basic Protocol 2</b>: Performing genome-wide genotyping of tandem repeats</p><p><b>Basic Protocol 3</b>: Discovering de novo tandem repeat expansions</p><p><b>Support Protocol 2</b>: Compiling ExpansionHunter Denovo from source code and generating STR profiles</p>","PeriodicalId":93970,"journal":{"name":"Current protocols","volume":"4 11","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-11-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cpz1.70010","citationCount":"0","resultStr":"{\"title\":\"Analysis of Tandem Repeats in Short-Read Sequencing Data: From Genotyping Known Pathogenic Repeats to Discovering Novel Expansions\",\"authors\":\"Andreas Halman, Andrew Lonsdale, Alicia Oshlack\",\"doi\":\"10.1002/cpz1.70010\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Short tandem repeats (STRs) and variable-number tandem repeats (VNTRs) are repetitive genomic sequences seen widely throughout the genome. These repeat expansions are currently known to cause ∼60 diseases, with expansions in new loci linked to rare diseases continuing to be discovered. Genome sequencing is an important tool for detecting disease-causing variants and several computational tools have been developed to analyze tandem repeats from genomic data, enabling the genotyping and the identification of expanded alleles. However, guidelines for conducting the analysis of these repeats and, more importantly, for assessing the findings are lacking. Understanding the tools and their technical limitations is important for accurately interpreting the results. This article provides detailed, step-by-step instructions for three key use cases in STR analysis from short-read genome sequencing data, which are also applicable to smaller VNTRs. First, it demonstrates an approach for genotyping known pathogenic loci and the identification of clinically significant expansions. Second, we offer guidance on defining tandem repeat loci and conducting genome-wide genotyping studies, which is also applicable to diploid organisms other than humans. Third, instructions are provided on how to find novel expansions at loci not previously known to be associated with disease, aiding in the discovery of new pathogenic loci. Moreover, we introduce the use of newly-developed helper tools that enable a complete and streamlined tandem repeat analysis protocol by addressing the gaps in current methods. All three protocols are compatible with human hg19, hg38, and the latest telomere-to-telomere (hs1) reference genomes. Additionally, this protocol provides an overview and discussion on how to interpret genotyping results. © 2024 The Author(s). Current Protocols published by Wiley Periodicals LLC.</p><p><b>Basic Protocol 1</b>: Genotyping known pathogenic tandem repeat loci</p><p><b>Alternate Protocol</b>: Genotyping known pathogenic tandem repeat loci with STRipy</p><p><b>Support Protocol 1</b>: Installation of tools and ExpansionHunter catalog modification</p><p><b>Basic Protocol 2</b>: Performing genome-wide genotyping of tandem repeats</p><p><b>Basic Protocol 3</b>: Discovering de novo tandem repeat expansions</p><p><b>Support Protocol 2</b>: Compiling ExpansionHunter Denovo from source code and generating STR profiles</p>\",\"PeriodicalId\":93970,\"journal\":{\"name\":\"Current protocols\",\"volume\":\"4 11\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-11-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cpz1.70010\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current protocols\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cpz1.70010\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current protocols","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cpz1.70010","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Analysis of Tandem Repeats in Short-Read Sequencing Data: From Genotyping Known Pathogenic Repeats to Discovering Novel Expansions
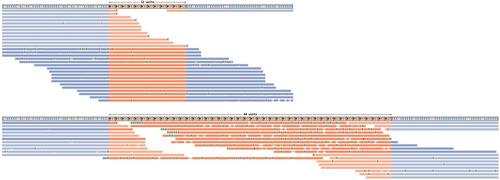
Short tandem repeats (STRs) and variable-number tandem repeats (VNTRs) are repetitive genomic sequences seen widely throughout the genome. These repeat expansions are currently known to cause ∼60 diseases, with expansions in new loci linked to rare diseases continuing to be discovered. Genome sequencing is an important tool for detecting disease-causing variants and several computational tools have been developed to analyze tandem repeats from genomic data, enabling the genotyping and the identification of expanded alleles. However, guidelines for conducting the analysis of these repeats and, more importantly, for assessing the findings are lacking. Understanding the tools and their technical limitations is important for accurately interpreting the results. This article provides detailed, step-by-step instructions for three key use cases in STR analysis from short-read genome sequencing data, which are also applicable to smaller VNTRs. First, it demonstrates an approach for genotyping known pathogenic loci and the identification of clinically significant expansions. Second, we offer guidance on defining tandem repeat loci and conducting genome-wide genotyping studies, which is also applicable to diploid organisms other than humans. Third, instructions are provided on how to find novel expansions at loci not previously known to be associated with disease, aiding in the discovery of new pathogenic loci. Moreover, we introduce the use of newly-developed helper tools that enable a complete and streamlined tandem repeat analysis protocol by addressing the gaps in current methods. All three protocols are compatible with human hg19, hg38, and the latest telomere-to-telomere (hs1) reference genomes. Additionally, this protocol provides an overview and discussion on how to interpret genotyping results. © 2024 The Author(s). Current Protocols published by Wiley Periodicals LLC.
Basic Protocol 1: Genotyping known pathogenic tandem repeat loci
Alternate Protocol: Genotyping known pathogenic tandem repeat loci with STRipy
Support Protocol 1: Installation of tools and ExpansionHunter catalog modification
Basic Protocol 2: Performing genome-wide genotyping of tandem repeats
Basic Protocol 3: Discovering de novo tandem repeat expansions
Support Protocol 2: Compiling ExpansionHunter Denovo from source code and generating STR profiles

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


