{"title":"两个丙炔基之间自反应的动力学和机理研究","authors":"Tien V. Pham, Nghia T. Nguyen, Tran Thu Huong","doi":"10.1007/s00894-024-06191-w","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The propargyl radical plays a critical role in various chemical processes, including hydrocarbon combustion, flame synthesis, and interstellar chemistry. Its unique stability arises from the delocalization of π-electrons, allowing it to participate in a wide range of reactions despite being a radical. The self-reaction of propargyl radicals is a fundamental step in synthesizing polycyclic aromatic hydrocarbons. In this work, therefore, a computational study into the C<sub>3</sub>H<sub>3</sub> + C<sub>3</sub>H<sub>3</sub> potential energy surface has been carefully characterized. The calculated results indicate that the reaction can occur by H-abstraction or addition of two propargyl radicals together. The H-abstraction mechanism can create the products P3 (H<sub>2</sub>CCC + H<sub>3</sub>CCCH) and P4 (H<sub>2</sub>CCCH<sub>2</sub> + HCCCH) but the energy barriers of the two H-abstraction channels are very high (from 12 to 22 kcal/mol). In contrast, the addition mechanism of two propargyl radicals forming the intermediates (I<sub>1</sub>, I<sub>5</sub>, I<sub>12</sub>) and the bimolecular products (P1, P2, P7, P11, P12) are dominant. Among the bimolecular products, the P11 (C<sub>6</sub>H<sub>4</sub> + H<sub>2</sub>) product is the most energetically favorable, and the channel leading to this product is also the most preferred path compared to all other paths throughout the PES. The calculated enthalpy changes of various reaction paths in this study are in good agreement with the available literature data, indicating that the CCSD(T) method is suitable for the title reaction. The overall rate constant of the reaction depends on both temperature and pressure, reducing with temperature but rising with pressure. The calculated results agree closely with the available experimental values and previous calculated data. Thus, it can be affirmed that in addition to the CASPT2 method as applied in the study of Georgievskii et al. (<i>Phys. Chem. Chem. Phys., 2007, 9, 4259–4268</i>), the CCSD(T) method is also very good for the self-reaction of two propargyl radicals.</p><h3>Methods</h3><p>The M06-2X and CCSD(T) methods with the aug-cc-pVTZ basis set were used to optimize and calculate single-point energies for all species of the reaction. The bimolecular rate constants of the dominant reaction paths were predicted in the temperature and pressure ranges of 300–1800 K and 0 – 76,000 Torr, respectively, using the VTST and RRKM models with Eckart tunneling correction for the H-shift steps.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 12","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-11-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A kinetic and mechanistic study of the self-reaction between two propargyl radicals\",\"authors\":\"Tien V. Pham, Nghia T. Nguyen, Tran Thu Huong\",\"doi\":\"10.1007/s00894-024-06191-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>The propargyl radical plays a critical role in various chemical processes, including hydrocarbon combustion, flame synthesis, and interstellar chemistry. Its unique stability arises from the delocalization of π-electrons, allowing it to participate in a wide range of reactions despite being a radical. The self-reaction of propargyl radicals is a fundamental step in synthesizing polycyclic aromatic hydrocarbons. In this work, therefore, a computational study into the C<sub>3</sub>H<sub>3</sub> + C<sub>3</sub>H<sub>3</sub> potential energy surface has been carefully characterized. The calculated results indicate that the reaction can occur by H-abstraction or addition of two propargyl radicals together. The H-abstraction mechanism can create the products P3 (H<sub>2</sub>CCC + H<sub>3</sub>CCCH) and P4 (H<sub>2</sub>CCCH<sub>2</sub> + HCCCH) but the energy barriers of the two H-abstraction channels are very high (from 12 to 22 kcal/mol). In contrast, the addition mechanism of two propargyl radicals forming the intermediates (I<sub>1</sub>, I<sub>5</sub>, I<sub>12</sub>) and the bimolecular products (P1, P2, P7, P11, P12) are dominant. Among the bimolecular products, the P11 (C<sub>6</sub>H<sub>4</sub> + H<sub>2</sub>) product is the most energetically favorable, and the channel leading to this product is also the most preferred path compared to all other paths throughout the PES. The calculated enthalpy changes of various reaction paths in this study are in good agreement with the available literature data, indicating that the CCSD(T) method is suitable for the title reaction. The overall rate constant of the reaction depends on both temperature and pressure, reducing with temperature but rising with pressure. The calculated results agree closely with the available experimental values and previous calculated data. Thus, it can be affirmed that in addition to the CASPT2 method as applied in the study of Georgievskii et al. (<i>Phys. Chem. Chem. Phys., 2007, 9, 4259–4268</i>), the CCSD(T) method is also very good for the self-reaction of two propargyl radicals.</p><h3>Methods</h3><p>The M06-2X and CCSD(T) methods with the aug-cc-pVTZ basis set were used to optimize and calculate single-point energies for all species of the reaction. The bimolecular rate constants of the dominant reaction paths were predicted in the temperature and pressure ranges of 300–1800 K and 0 – 76,000 Torr, respectively, using the VTST and RRKM models with Eckart tunneling correction for the H-shift steps.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"30 12\",\"pages\":\"\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2024-11-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-024-06191-w\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06191-w","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
A kinetic and mechanistic study of the self-reaction between two propargyl radicals
Context
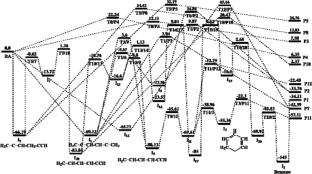
The propargyl radical plays a critical role in various chemical processes, including hydrocarbon combustion, flame synthesis, and interstellar chemistry. Its unique stability arises from the delocalization of π-electrons, allowing it to participate in a wide range of reactions despite being a radical. The self-reaction of propargyl radicals is a fundamental step in synthesizing polycyclic aromatic hydrocarbons. In this work, therefore, a computational study into the C3H3 + C3H3 potential energy surface has been carefully characterized. The calculated results indicate that the reaction can occur by H-abstraction or addition of two propargyl radicals together. The H-abstraction mechanism can create the products P3 (H2CCC + H3CCCH) and P4 (H2CCCH2 + HCCCH) but the energy barriers of the two H-abstraction channels are very high (from 12 to 22 kcal/mol). In contrast, the addition mechanism of two propargyl radicals forming the intermediates (I1, I5, I12) and the bimolecular products (P1, P2, P7, P11, P12) are dominant. Among the bimolecular products, the P11 (C6H4 + H2) product is the most energetically favorable, and the channel leading to this product is also the most preferred path compared to all other paths throughout the PES. The calculated enthalpy changes of various reaction paths in this study are in good agreement with the available literature data, indicating that the CCSD(T) method is suitable for the title reaction. The overall rate constant of the reaction depends on both temperature and pressure, reducing with temperature but rising with pressure. The calculated results agree closely with the available experimental values and previous calculated data. Thus, it can be affirmed that in addition to the CASPT2 method as applied in the study of Georgievskii et al. (Phys. Chem. Chem. Phys., 2007, 9, 4259–4268), the CCSD(T) method is also very good for the self-reaction of two propargyl radicals.
Methods
The M06-2X and CCSD(T) methods with the aug-cc-pVTZ basis set were used to optimize and calculate single-point energies for all species of the reaction. The bimolecular rate constants of the dominant reaction paths were predicted in the temperature and pressure ranges of 300–1800 K and 0 – 76,000 Torr, respectively, using the VTST and RRKM models with Eckart tunneling correction for the H-shift steps.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


