Gabriel T Galdino, Olivier Mailhot, Rafael Najmanovich
{"title":"通过对复杂结构的定量动态分析了解和预测配体在μ-阿片受体中的功效","authors":"Gabriel T Galdino, Olivier Mailhot, Rafael Najmanovich","doi":"10.1021/acs.jcim.4c00788","DOIUrl":null,"url":null,"abstract":"<p><p>The μ-opioid receptor (MOR) is a G-protein coupled receptor involved in nociception and the primary target of opioid drugs. Understanding the relationships among the ligand structure, receptor dynamics, and efficacy in activating MOR is crucial for drug discovery and development. Here, we use coarse-grained normal-mode analysis to predict ligand-induced changes in receptor dynamics with the Quantitative Dynamics Activity Relationship (QDAR) DynaSig-ML methodology, training a LASSO regression model on the entropic signatures (ESs) computed from ligand-receptor complexes. We train and validate the methodology using a data set of 179 MOR ligands with experimentally measured efficacies split into strictly chemically different cross-validation sets. By analyzing the coefficients of the ES LASSO model, we identified key residues involved in MOR activation, several of which have mutational data supporting their role in MOR activation. Additionally, we explored a contact-only LASSO model based on ligand-protein interactions. While the model showed predictive power, it failed at predicting efficacy for ligands with low structural similarity to the training set, emphasizing the importance of receptor dynamics for predicting ligand-induced receptor activation. Moreover, the low computational cost of our approach, at 3 CPU s per ligand-receptor complex, opens the door to its application in large-scale virtual screening contexts. Our work contributes to a better understanding of dynamics-function relationships in the μ-opioid receptor and provides a framework for predicting ligand efficacy based on ligand-induced changes in receptor dynamics.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"8549-8561"},"PeriodicalIF":5.6000,"publicationDate":"2024-11-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Understanding and Predicting Ligand Efficacy in the μ-Opioid Receptor through Quantitative Dynamical Analysis of Complex Structures.\",\"authors\":\"Gabriel T Galdino, Olivier Mailhot, Rafael Najmanovich\",\"doi\":\"10.1021/acs.jcim.4c00788\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The μ-opioid receptor (MOR) is a G-protein coupled receptor involved in nociception and the primary target of opioid drugs. Understanding the relationships among the ligand structure, receptor dynamics, and efficacy in activating MOR is crucial for drug discovery and development. Here, we use coarse-grained normal-mode analysis to predict ligand-induced changes in receptor dynamics with the Quantitative Dynamics Activity Relationship (QDAR) DynaSig-ML methodology, training a LASSO regression model on the entropic signatures (ESs) computed from ligand-receptor complexes. We train and validate the methodology using a data set of 179 MOR ligands with experimentally measured efficacies split into strictly chemically different cross-validation sets. By analyzing the coefficients of the ES LASSO model, we identified key residues involved in MOR activation, several of which have mutational data supporting their role in MOR activation. Additionally, we explored a contact-only LASSO model based on ligand-protein interactions. While the model showed predictive power, it failed at predicting efficacy for ligands with low structural similarity to the training set, emphasizing the importance of receptor dynamics for predicting ligand-induced receptor activation. Moreover, the low computational cost of our approach, at 3 CPU s per ligand-receptor complex, opens the door to its application in large-scale virtual screening contexts. Our work contributes to a better understanding of dynamics-function relationships in the μ-opioid receptor and provides a framework for predicting ligand efficacy based on ligand-induced changes in receptor dynamics.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"8549-8561\"},\"PeriodicalIF\":5.6000,\"publicationDate\":\"2024-11-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.4c00788\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00788","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
摘要
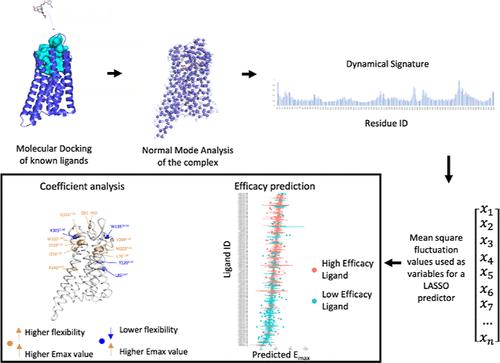
μ-阿片受体(MOR)是一种参与痛觉的G蛋白偶联受体,也是阿片类药物的主要靶点。了解配体结构、受体动力学和激活 MOR 的功效之间的关系对于药物发现和开发至关重要。在这里,我们利用粗粒度正态模式分析预测配体诱导的受体动力学变化,采用定量动力学活性关系(QDAR)DynaSig-ML 方法,在配体-受体复合物计算出的熵特征(ES)上训练 LASSO 回归模型。我们使用由 179 种 MOR 配体组成的数据集对该方法进行了训练和验证,这些配体的药效是通过实验测得的,并分成了化学性质严格不同的交叉验证集。通过分析 ES LASSO 模型的系数,我们确定了参与 MOR 激活的关键残基,其中几个残基的突变数据支持它们在 MOR 激活中的作用。此外,我们还探索了基于配体与蛋白质相互作用的纯接触 LASSO 模型。虽然该模型显示出了预测能力,但它无法预测与训练集结构相似度较低的配体的药效,这强调了受体动力学对预测配体诱导的受体激活的重要性。此外,我们的方法计算成本低,每个配体-受体复合物只需 3 CPU s,这为其在大规模虚拟筛选中的应用打开了大门。我们的工作有助于更好地理解μ-阿片受体的动力学-功能关系,并为根据配体诱导的受体动力学变化预测配体功效提供了一个框架。
Understanding and Predicting Ligand Efficacy in the μ-Opioid Receptor through Quantitative Dynamical Analysis of Complex Structures.
The μ-opioid receptor (MOR) is a G-protein coupled receptor involved in nociception and the primary target of opioid drugs. Understanding the relationships among the ligand structure, receptor dynamics, and efficacy in activating MOR is crucial for drug discovery and development. Here, we use coarse-grained normal-mode analysis to predict ligand-induced changes in receptor dynamics with the Quantitative Dynamics Activity Relationship (QDAR) DynaSig-ML methodology, training a LASSO regression model on the entropic signatures (ESs) computed from ligand-receptor complexes. We train and validate the methodology using a data set of 179 MOR ligands with experimentally measured efficacies split into strictly chemically different cross-validation sets. By analyzing the coefficients of the ES LASSO model, we identified key residues involved in MOR activation, several of which have mutational data supporting their role in MOR activation. Additionally, we explored a contact-only LASSO model based on ligand-protein interactions. While the model showed predictive power, it failed at predicting efficacy for ligands with low structural similarity to the training set, emphasizing the importance of receptor dynamics for predicting ligand-induced receptor activation. Moreover, the low computational cost of our approach, at 3 CPU s per ligand-receptor complex, opens the door to its application in large-scale virtual screening contexts. Our work contributes to a better understanding of dynamics-function relationships in the μ-opioid receptor and provides a framework for predicting ligand efficacy based on ligand-induced changes in receptor dynamics.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


