{"title":"改善高能吡嗪及其 N-氧化物的特性。","authors":"Dmitry V. Khakimov, Tatyana S. Pivina","doi":"10.1007/s00894-024-06186-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Based on the methods of quantum chemistry and atom–atom potentials, the molecular and crystal structure of a number of high-energy pyrazines was modeled: unsubstituted diazines, as well as fully nitrated 1,4-diazabenzenes, their oxides and polymorphs. The enthalpies of formation, densities of molecular crystals, and some performance characteristics of these compounds were determined. The parameters of decomposition of substances were estimated. It has been established that tetranitropyrazine-1,4-dioxide has maximum energy content and excellent performance characteristics, which determine the prospects for using this compound as a high-energy one in the considered series of compounds.</p><h3>Methods</h3><p>In this work, DFT calculations were conducted through the software Gaussian 09 using B3LYP functional with basis set aug-cc-PVDZ and the Grimme dispersion correction D2. For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP and enthalpies of formation in gas phase were assessed by G3B3.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 11","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Towards improving the characteristics of high-energy pyrazines and their N-oxides\",\"authors\":\"Dmitry V. Khakimov, Tatyana S. Pivina\",\"doi\":\"10.1007/s00894-024-06186-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>Based on the methods of quantum chemistry and atom–atom potentials, the molecular and crystal structure of a number of high-energy pyrazines was modeled: unsubstituted diazines, as well as fully nitrated 1,4-diazabenzenes, their oxides and polymorphs. The enthalpies of formation, densities of molecular crystals, and some performance characteristics of these compounds were determined. The parameters of decomposition of substances were estimated. It has been established that tetranitropyrazine-1,4-dioxide has maximum energy content and excellent performance characteristics, which determine the prospects for using this compound as a high-energy one in the considered series of compounds.</p><h3>Methods</h3><p>In this work, DFT calculations were conducted through the software Gaussian 09 using B3LYP functional with basis set aug-cc-PVDZ and the Grimme dispersion correction D2. For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP and enthalpies of formation in gas phase were assessed by G3B3.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"30 11\",\"pages\":\"\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-024-06186-7\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06186-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Towards improving the characteristics of high-energy pyrazines and their N-oxides
Context
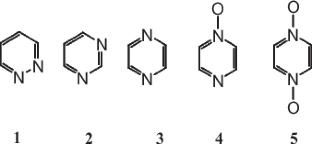
Based on the methods of quantum chemistry and atom–atom potentials, the molecular and crystal structure of a number of high-energy pyrazines was modeled: unsubstituted diazines, as well as fully nitrated 1,4-diazabenzenes, their oxides and polymorphs. The enthalpies of formation, densities of molecular crystals, and some performance characteristics of these compounds were determined. The parameters of decomposition of substances were estimated. It has been established that tetranitropyrazine-1,4-dioxide has maximum energy content and excellent performance characteristics, which determine the prospects for using this compound as a high-energy one in the considered series of compounds.
Methods
In this work, DFT calculations were conducted through the software Gaussian 09 using B3LYP functional with basis set aug-cc-PVDZ and the Grimme dispersion correction D2. For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP and enthalpies of formation in gas phase were assessed by G3B3.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


