Gustav Olanders, Giulia Testa, Alessandro Tibo, Eva Nittinger, Christian Tyrchan
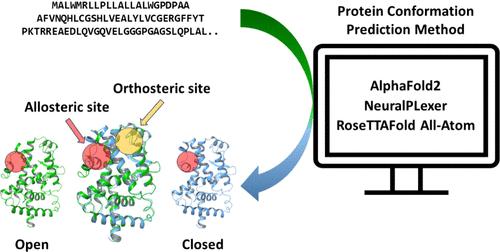
{"title":"深度学习的挑战:配体诱导异位和正位构象变化的蛋白质结构预测。","authors":"Gustav Olanders, Giulia Testa, Alessandro Tibo, Eva Nittinger, Christian Tyrchan","doi":"10.1021/acs.jcim.4c01475","DOIUrl":null,"url":null,"abstract":"<p><p>In the realm of biomedical research, understanding the intricate structure of proteins is crucial, as these structures determine how proteins function within our bodies and interact with potential drugs. Traditionally, methods like X-ray crystallography and cryo-electron microscopy have been used to unravel these structures, but they are often challenging, time-consuming and costly. Recently, a breakthrough in computational biology has emerged with the development of deep learning algorithms capable of predicting protein structures based on their amino acid sequences (Jumper, J., et al. <i>Nature</i> <b>2021</b>, <i>596</i>, 583. Lane, T. J. <i>Nature Methods</i> <b>2023</b>, <i>20</i>, 170. Kryshtafovych, A., et al. <i>Proteins: Structure, Function and Bioinformatics</i> <b>2021</b>, <i>89</i>, 1607). This study focuses on predicting the dynamic changes that proteins undergo upon ligand binding, specifically when they bind to allosteric sites, i.e. a pocket different from the active site. Allosteric modulators are particularly important for drug discovery, as they open new avenues for designing drugs that can target proteins more effectively and with fewer side effects (Nussinov, R.; Tsai, C. J. <i>Cell</i> <b>2013</b>, <i>153</i>, 293). To study this, we curated a data set of 578 X-ray structures comprised of proteins displaying orthosteric and allosteric binding as well as a general framework to evaluate deep learning-based structure prediction methods. Our findings demonstrate the potential and current limitations of deep learning methods, such as AlphaFold2 (Jumper, J., et al. <i>Nature</i> <b>2021</b>, <i>596</i>, 583), NeuralPLexer (Qiao, Z., et al. <i>Nat Mach Intell</i> <b>2024</b>, <i>6</i>, 195), and RoseTTAFold All-Atom (Krishna, R., et al. <i>Science</i> <b>2024</b>, <i>384</i>, eadl2528) to predict not just static protein structures but also the dynamic conformational changes. Herein we show that predicting the allosteric induce-fit conformation still poses a challenge to deep learning methods as they more accurately predict the orthosteric bound conformation compared to the allosteric induce fit conformation. For AlphaFold2, we observed that conformational diversity, and sampling between the apo and holo state could be increased by modifying the MSA depth, but this did not enhance the ability to generate conformations close to the allosteric induced-fit conformation. To further support advancements in protein structure prediction field, the curated data set and evaluation framework are made publicly available.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":""},"PeriodicalIF":5.6000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Challenge for Deep Learning: Protein Structure Prediction of Ligand-Induced Conformational Changes at Allosteric and Orthosteric Sites.\",\"authors\":\"Gustav Olanders, Giulia Testa, Alessandro Tibo, Eva Nittinger, Christian Tyrchan\",\"doi\":\"10.1021/acs.jcim.4c01475\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>In the realm of biomedical research, understanding the intricate structure of proteins is crucial, as these structures determine how proteins function within our bodies and interact with potential drugs. Traditionally, methods like X-ray crystallography and cryo-electron microscopy have been used to unravel these structures, but they are often challenging, time-consuming and costly. Recently, a breakthrough in computational biology has emerged with the development of deep learning algorithms capable of predicting protein structures based on their amino acid sequences (Jumper, J., et al. <i>Nature</i> <b>2021</b>, <i>596</i>, 583. Lane, T. J. <i>Nature Methods</i> <b>2023</b>, <i>20</i>, 170. Kryshtafovych, A., et al. <i>Proteins: Structure, Function and Bioinformatics</i> <b>2021</b>, <i>89</i>, 1607). This study focuses on predicting the dynamic changes that proteins undergo upon ligand binding, specifically when they bind to allosteric sites, i.e. a pocket different from the active site. Allosteric modulators are particularly important for drug discovery, as they open new avenues for designing drugs that can target proteins more effectively and with fewer side effects (Nussinov, R.; Tsai, C. J. <i>Cell</i> <b>2013</b>, <i>153</i>, 293). To study this, we curated a data set of 578 X-ray structures comprised of proteins displaying orthosteric and allosteric binding as well as a general framework to evaluate deep learning-based structure prediction methods. Our findings demonstrate the potential and current limitations of deep learning methods, such as AlphaFold2 (Jumper, J., et al. <i>Nature</i> <b>2021</b>, <i>596</i>, 583), NeuralPLexer (Qiao, Z., et al. <i>Nat Mach Intell</i> <b>2024</b>, <i>6</i>, 195), and RoseTTAFold All-Atom (Krishna, R., et al. <i>Science</i> <b>2024</b>, <i>384</i>, eadl2528) to predict not just static protein structures but also the dynamic conformational changes. Herein we show that predicting the allosteric induce-fit conformation still poses a challenge to deep learning methods as they more accurately predict the orthosteric bound conformation compared to the allosteric induce fit conformation. For AlphaFold2, we observed that conformational diversity, and sampling between the apo and holo state could be increased by modifying the MSA depth, but this did not enhance the ability to generate conformations close to the allosteric induced-fit conformation. To further support advancements in protein structure prediction field, the curated data set and evaluation framework are made publicly available.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.6000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.4c01475\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c01475","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
摘要
在生物医学研究领域,了解蛋白质的复杂结构至关重要,因为这些结构决定了蛋白质在人体内的功能以及与潜在药物的相互作用。传统上,人们使用 X 射线晶体学和冷冻电镜等方法来揭示这些结构,但这些方法往往具有挑战性、耗时且成本高昂。最近,随着能够根据氨基酸序列预测蛋白质结构的深度学习算法的发展,计算生物学出现了突破性进展(Jumper, J., et al. Nature 2021, 596, 583.Lane, T. J. Nature Methods 2023, 20, 170.Kryshtafovych, A., et al. Proteins: Structure, Function and Bioinformatics 2021, 89, 1607)。这项研究的重点是预测蛋白质与配体结合后发生的动态变化,特别是当配体与异构位点(即不同于活性位点的口袋)结合时。异位调节剂对药物发现尤为重要,因为它们为设计能更有效地靶向蛋白质且副作用更小的药物开辟了新途径(Nussinov, R.; Tsai, C. J. Cell 2013, 153, 293)。为了研究这一点,我们整理了一个包含 578 个 X 射线结构的数据集,这些数据集由显示正交和异位结合的蛋白质组成,同时还整理了一个通用框架,用于评估基于深度学习的结构预测方法。我们的研究结果表明了深度学习方法的潜力和目前的局限性,如 AlphaFold2(Jumper, J., et al. Nature 2021, 596, 583)、NeuralPLexer(Qiao, Z., et al. Nat Mach Intell 2024, 6, 195)和 RoseTTAFold All-Atom(Krishna, R., et al. Science 2024, 384, eadl2528),它们不仅能预测静态蛋白质结构,还能预测动态构象变化。在这里,我们发现预测异构诱导拟合构象仍然是深度学习方法面临的挑战,因为与异构诱导拟合构象相比,深度学习方法能更准确地预测正交结合构象。对于 AlphaFold2,我们观察到构象多样性以及 apo 和 holo 状态之间的采样可以通过修改 MSA 深度来增加,但这并没有提高生成接近于异生诱导拟合构象的能力。为了进一步支持蛋白质结构预测领域的进步,我们公开了经整理的数据集和评估框架。
Challenge for Deep Learning: Protein Structure Prediction of Ligand-Induced Conformational Changes at Allosteric and Orthosteric Sites.
In the realm of biomedical research, understanding the intricate structure of proteins is crucial, as these structures determine how proteins function within our bodies and interact with potential drugs. Traditionally, methods like X-ray crystallography and cryo-electron microscopy have been used to unravel these structures, but they are often challenging, time-consuming and costly. Recently, a breakthrough in computational biology has emerged with the development of deep learning algorithms capable of predicting protein structures based on their amino acid sequences (Jumper, J., et al. Nature2021, 596, 583. Lane, T. J. Nature Methods2023, 20, 170. Kryshtafovych, A., et al. Proteins: Structure, Function and Bioinformatics2021, 89, 1607). This study focuses on predicting the dynamic changes that proteins undergo upon ligand binding, specifically when they bind to allosteric sites, i.e. a pocket different from the active site. Allosteric modulators are particularly important for drug discovery, as they open new avenues for designing drugs that can target proteins more effectively and with fewer side effects (Nussinov, R.; Tsai, C. J. Cell2013, 153, 293). To study this, we curated a data set of 578 X-ray structures comprised of proteins displaying orthosteric and allosteric binding as well as a general framework to evaluate deep learning-based structure prediction methods. Our findings demonstrate the potential and current limitations of deep learning methods, such as AlphaFold2 (Jumper, J., et al. Nature2021, 596, 583), NeuralPLexer (Qiao, Z., et al. Nat Mach Intell2024, 6, 195), and RoseTTAFold All-Atom (Krishna, R., et al. Science2024, 384, eadl2528) to predict not just static protein structures but also the dynamic conformational changes. Herein we show that predicting the allosteric induce-fit conformation still poses a challenge to deep learning methods as they more accurately predict the orthosteric bound conformation compared to the allosteric induce fit conformation. For AlphaFold2, we observed that conformational diversity, and sampling between the apo and holo state could be increased by modifying the MSA depth, but this did not enhance the ability to generate conformations close to the allosteric induced-fit conformation. To further support advancements in protein structure prediction field, the curated data set and evaluation framework are made publicly available.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


