MOFSynth:用于 MOFs 合成可能性预测的计算工具
IF 5.6
2区 化学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
在过去十年中,主要由于计算机能力的进步,材料的高通量计算研究大幅增加,并引起了人们的极大兴趣。在金属有机框架(MOFs)领域,已经在硅学中设计了超过一百万个假定的 MOFs,但其中只有一小部分得到了合成。为了验证计算-假想结果并加快该领域的进展,迫切需要区分出更有可能合成用于实际应用的 MOF。本研究对 MOF 的合成可能性进行了全面调查,采用了一种新颖的计算方法,该方法基于 MOF 结构中的链接构象与其孤立的自由气体状态之间在能量和几何形状上的差异,因为这两者已被证明是影响 MOF 合成的关键因素。我们的用户友好型工具可简化可合成性评估,只需最低限度的计算化学专业知识。通过解构 QMOF、CoRE MOF 和 ToBaCCo 等数据库中的 40,000 多种 MOF,我们分析了定义 MOF 单元池内链节应变的关键参数。结果表明,QMOF 和 CoRE MOF 包含更多具有合成前景的候选材料,而 ToBaCCo 则由于材料未优化而显示出相对较差的可合成性。通过大量分析,我们确定了高可合成性 MOF 的最佳链接器候选材料。高皮尔逊系数和斯皮尔曼系数证实了数据库中能量分布的一致趋势,这表明省略优化计算的潜力巨大,可显著降低计算成本。这项研究强调了连接体变形和能量差异的重要性,加深了我们对 MOF 研究中合成可及性的理解,为该领域未来的发展提供了宝贵的见解。本文章由计算机程序翻译,如有差异,请以英文原文为准。
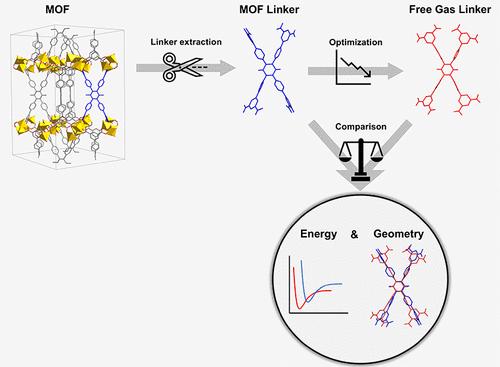
MOFSynth: A Computational Tool toward Synthetic Likelihood Predictions of MOFs
In the past decade, high-throughput computational studies of materials have increased significantly mainly due to advances in computer capabilities and have attracted a great deal of interest. In the field of metal–organic frameworks (MOFs), over a million hypothetical MOFs have been designed in silico, yet only a small fraction of these have been synthesized. For validating the computational-hypothetical results and accelerating the progress in the field, there is a pressing need for distinguishing MOFs that are more likely to be synthesized for real-life applications. This study presents a comprehensive investigation into the synthesizability likelihood of MOFs, utilizing a novel computational approach based on the disparities in energy and geometry between the linker conformation within the MOF structure and its isolated, free-gas state since both of these have been proven to be critical factors influencing MOF synthesis. Our user-friendly tool streamlines synthesizability evaluation, requiring minimal expertise in computational chemistry. By deconstructing over 40,000 MOFs from databases, including QMOF, CoRE MOF, and ToBaCCo, we analyze key parameters defining the linker strain within the MOF unit cell. Our results indicate that QMOF and CoRE MOF contain more promising candidates for synthesis, while ToBaCCo exhibits a relatively poor synthesizability likelihood due to unoptimized materials. Through extensive analysis, we identify optimal linker candidates for highly synthesizable MOFs. Consistent trends in energy distribution across databases that are confirmed by high Pearson and Spearman coefficients suggest the potential for omitting optimization calculations, significantly reducing computational costs. This study underscores the importance of linker deformation and energy disparities and enhances our understanding of synthetic accessibility in MOF research, offering valuable insights for future advancements in the field.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
9.80
自引率
10.70%
发文量
529
审稿时长
1.4 months
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


