Shailesh Kumar Panday, Arghya Chakravorty, Shan Zhao, Emil Alexov
{"title":"利用正则化超高斯泊松-波尔兹曼模型,从能量最小化结构中传递蛋白质的极性溶解自由能。","authors":"Shailesh Kumar Panday, Arghya Chakravorty, Shan Zhao, Emil Alexov","doi":"10.1002/jcc.27496","DOIUrl":null,"url":null,"abstract":"<p>The biomolecules interact with their partners in an aqueous media; thus, their solvation energy is an important thermodynamics quantity. In previous works (<i>J. Chem. Theory Comput. 14</i>(2): 1020–1032), we demonstrated that the Poisson–Boltzmann (PB) approach reproduces solvation energy calculated via thermodynamic integration (TI) protocol if the structures of proteins are kept rigid. However, proteins are not rigid bodies and computing their solvation energy must account for their flexibility. Typically, in the framework of PB calculations, this is done by collecting snapshots from molecular dynamics (MD) simulations, computing their solvation energies, and averaging to obtain the ensemble-averaged solvation energy, which is computationally demanding. To reduce the computational cost, we have proposed Gaussian/super-Gaussian-based methods for the dielectric function that use the atomic packing to deliver smooth dielectric function for the entire computational space, the protein and water phase, which allows the ensemble-averaged solvation energy to be computed from a single structure. One of the technical difficulties associated with the smooth dielectric function presentation with respect to polar solvation energy is the absence of a dielectric border between the protein and water where induced charges should be positioned. This motivated the present work, where we report a super-Gaussian regularized Poisson–Boltzmann method and use it for computing the polar solvation energy from single energy minimized structures and assess its ability to reproduce the ensemble-averaged polar solvation on a dataset of 74 high-resolution monomeric proteins.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-10-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27496","citationCount":"0","resultStr":"{\"title\":\"On delivering polar solvation free energy of proteins from energy minimized structures using a regularized super-Gaussian Poisson–Boltzmann model\",\"authors\":\"Shailesh Kumar Panday, Arghya Chakravorty, Shan Zhao, Emil Alexov\",\"doi\":\"10.1002/jcc.27496\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The biomolecules interact with their partners in an aqueous media; thus, their solvation energy is an important thermodynamics quantity. In previous works (<i>J. Chem. Theory Comput. 14</i>(2): 1020–1032), we demonstrated that the Poisson–Boltzmann (PB) approach reproduces solvation energy calculated via thermodynamic integration (TI) protocol if the structures of proteins are kept rigid. However, proteins are not rigid bodies and computing their solvation energy must account for their flexibility. Typically, in the framework of PB calculations, this is done by collecting snapshots from molecular dynamics (MD) simulations, computing their solvation energies, and averaging to obtain the ensemble-averaged solvation energy, which is computationally demanding. To reduce the computational cost, we have proposed Gaussian/super-Gaussian-based methods for the dielectric function that use the atomic packing to deliver smooth dielectric function for the entire computational space, the protein and water phase, which allows the ensemble-averaged solvation energy to be computed from a single structure. One of the technical difficulties associated with the smooth dielectric function presentation with respect to polar solvation energy is the absence of a dielectric border between the protein and water where induced charges should be positioned. This motivated the present work, where we report a super-Gaussian regularized Poisson–Boltzmann method and use it for computing the polar solvation energy from single energy minimized structures and assess its ability to reproduce the ensemble-averaged polar solvation on a dataset of 74 high-resolution monomeric proteins.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-10-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27496\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27496\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27496","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
On delivering polar solvation free energy of proteins from energy minimized structures using a regularized super-Gaussian Poisson–Boltzmann model
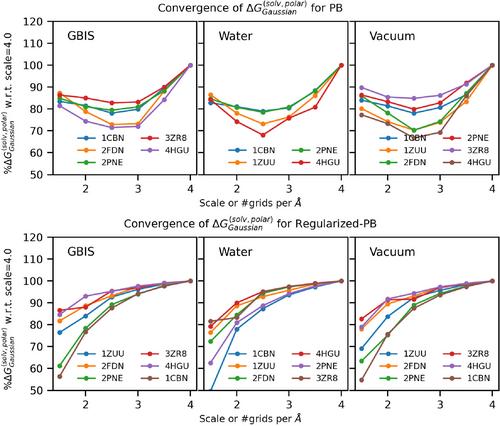
The biomolecules interact with their partners in an aqueous media; thus, their solvation energy is an important thermodynamics quantity. In previous works (J. Chem. Theory Comput. 14(2): 1020–1032), we demonstrated that the Poisson–Boltzmann (PB) approach reproduces solvation energy calculated via thermodynamic integration (TI) protocol if the structures of proteins are kept rigid. However, proteins are not rigid bodies and computing their solvation energy must account for their flexibility. Typically, in the framework of PB calculations, this is done by collecting snapshots from molecular dynamics (MD) simulations, computing their solvation energies, and averaging to obtain the ensemble-averaged solvation energy, which is computationally demanding. To reduce the computational cost, we have proposed Gaussian/super-Gaussian-based methods for the dielectric function that use the atomic packing to deliver smooth dielectric function for the entire computational space, the protein and water phase, which allows the ensemble-averaged solvation energy to be computed from a single structure. One of the technical difficulties associated with the smooth dielectric function presentation with respect to polar solvation energy is the absence of a dielectric border between the protein and water where induced charges should be positioned. This motivated the present work, where we report a super-Gaussian regularized Poisson–Boltzmann method and use it for computing the polar solvation energy from single energy minimized structures and assess its ability to reproduce the ensemble-averaged polar solvation on a dataset of 74 high-resolution monomeric proteins.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


