聚合物半金属(Si、Ge 和 As)和非金属(N 和 P)掺杂 C70-Fullerene 系统的最新进展:电子、动态行为和化疗对接与 ADMET 分析的比较
IF 2.1
3区 化学
Q3 CHEMISTRY, INORGANIC & NUCLEAR
引用次数: 0
摘要
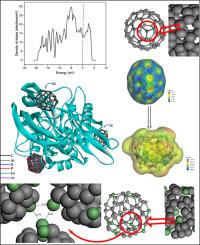
对基于碳原子的富勒烯系统进行了研究,并在考虑到 Si、N、P、Ge 和 As 等元素的情况下对其结构进行了改性,从而构建了掺杂改性系统。研究还考虑了合成路线调查,以获得与结构特性研究相关的进一步应用中的结构可用性。对这些设计系统的计算研究采用了最流行的密度泛函理论(DFT)方法。尤其是通过状态密度(DOS)光谱、分子静电位(MEP)和前沿分子轨道(FMOs)对所有系统的电子行为进行了研究,以便对能量较低的稳定性进行最佳比较,其中 C-fullerene 被预测为更稳定的候选物质,因为其 ∆E 值为 1.714 eV。分子动力学模拟(MD)被用来预测系统中原子在 100 ps 物理运动过程中的稳定性。为每个半金属原子(Si、Ge 和 As)链接了三重富勒烯环的聚合物模型,以预测稳定条件下的径向分布函数 (RDF) 和动态行为。计算得出的势能、动能和非键能等能量参数主要描述了模拟时间内系统运动的稳定性。为了预测所研究体系对乳腺癌靶蛋白(5NWH)的化疗作用,对改性掺杂体系进行了分子对接分析,通过活性位点结合亲和力评估抑制强度。结果发现,Si-fullerene 的估计结合亲和力(-12.73 kcal/mol)最为理想。通过比较药代动力学因素,对 ADMET 特性进行了估计,以便进一步进行类药物预测。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Recent perspective on polymeric Semimetal (Si, Ge and As) and nonmetal (N and P) doped C70-Fullerene system: Comparative electronic, dynamic behavior and chemotherapy docking with ADMET analysis
Fullerene system, based on carbon atoms, was studied and its structural modification was built with consideration of several elements such as Si, N, P, Ge, and As, where doped modified systems were constructed. Synthetic routes survey was considered to acquire the structures' availability in further applications related to structure properties investigation. The computational investigation of these designed systems was described using the most popular approach of density functional theory (DFT). Electronic behavior for all systems was studied especially through density of states (DOS) spectra, molecular electrostatic potential (MEP), and frontier molecular orbitals (FMOs) for best comparison towards stability with less energetic properties, where C-fullerene was predicted as a more stable candidate as its ∆E with value of 1.714 eV. Molecular dynamic simulation (MD) was considered to predict the stability of the atoms in the system during the physical movement at 100 ps. A polymeric model of triple Fullerene rings was linked for each Semimetal atom (Si, Ge, and As) to predict the radial distribution function (RDF) and dynamic behavior for stability conditions. The calculated energy parameters such as potential, kinetic, and non-bond energies mainly described the system movement stability during the simulation time. Molecular docking analysis of the modified doped systems was performed for chemotherapy prediction of the studied systems against breast cancer target protein (5NWH) to evaluate the inhibition strength through active sites binding affinity. The estimated binding affinity of Si-fullerene was found the most favorable result (-12.73 kcal/mol). ADMET properties were estimated for further drug-like prediction through comparative pharmacokinetic factors.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Organometallic Chemistry
化学-无机化学与核化学
CiteScore
4.40
自引率
8.70%
发文量
221
审稿时长
36 days
期刊介绍:
The Journal of Organometallic Chemistry targets original papers dealing with theoretical aspects, structural chemistry, synthesis, physical and chemical properties (including reaction mechanisms), and practical applications of organometallic compounds.
Organometallic compounds are defined as compounds that contain metal - carbon bonds. The term metal includes all alkali and alkaline earth metals, all transition metals and the lanthanides and actinides in the Periodic Table. Metalloids including the elements in Group 13 and the heavier members of the Groups 14 - 16 are also included. The term chemistry includes syntheses, characterizations and reaction chemistry of all such compounds. Research reports based on use of organometallic complexes in bioorganometallic chemistry, medicine, material sciences, homogeneous catalysis and energy conversion are also welcome.
The scope of the journal has been enlarged to encompass important research on organometallic complexes in bioorganometallic chemistry and material sciences, and of heavier main group elements in organometallic chemistry. The journal also publishes review articles, short communications and notes.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


