铂纳米团簇氢吸附能的机器学习预测:SOAP 描述子比较研究
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
金属材料中的氢结合能在储氢和电催化氢进化反应中具有重要意义。本研究通过第一性原理计算获得了不同尺寸铂纳米团簇吸附氢气的数据集(超过 9000 个数据)。数据分析表明,氢与铂的结合强度与吸附位点的局部结构密切相关。铂与氢之间的距离和纳米团簇的大小等局部特征与原子位置平滑重叠描述符相辅相成,通过机器学习方法拟合并预测氢在不同铂纳米结构上的吸附能。使用高斯过程回归(GPR)和随机森林回归器(RFR)构建氢结合能预测模型,发现使用修改后的描述符,测试集的 R2 从 0.63 提高到 0.78。将其应用到其他纳米团簇中,预测模型的 MAE 为 0.08 eV,显示出氢吸附能的高精度。我们的模型可以很容易地扩展到其他材料的氢吸附能预测中,而且计算成本和精度都不高,这将有助于高性能催化剂的结构设计。本文章由计算机程序翻译,如有差异,请以英文原文为准。
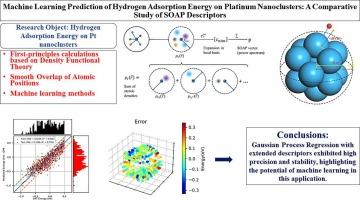
Machine learning prediction of hydrogen adsorption energy on platinum nanoclusters: A comparative study of SOAP descriptors
Hydrogen binding energy in metal materials is of high significance in the hydrogen storage as well as the hydrogen evolution reaction of electrocatalysis. In this work, the datasets (more than 9000 data) of hydrogen adsorbed on Pt nanoclusters with different sizes are obtained by first-principles calculations. Data analysis shows that the binding strength of hydrogen with Pt is closely relevant to the local structures of the adsorption sites. The local features of the distance between the platinum and hydrogen and the size of the nanoclusters are supplemented to the Smooth Overlap of Atomic Positions descriptors to fit and predict the adsorption energies of hydrogen on different Pt nano-structures by performing the machine learning method. Gaussian Process Regression (GPR) and Random Forest Regressor (RFR) are used to construct the prediction model of hydrogen binding energies and it is found the R2 of test set is improved from 0.63 to 0.78 with modified descriptors. By applying it into other nanoclusters, the MAE of the prediction model is 0.08 eV, which exhibits high accuracy of the hydrogen adsorption energy. Our model can be easily extended to the prediction of hydrogen adsorption energy of other materials with affordable computational cost and accuracy, which would be helpful for the structural design of high-performance catalysts.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


