{"title":"整合单细胞 RNA 测序数据,解读巨噬细胞在不同器官和疾病中的异质性和功能","authors":"Xinjie Xu, Zichen Wu, Zhiwei Zeng, Jiaying Cao, Liang Chen","doi":"10.1002/ctd2.70005","DOIUrl":null,"url":null,"abstract":"<p>Macrophages are essential components of the innate immune system, playing crucial roles in host defence, immune surveillance and tissue repair processes. Developmentally, macrophages can be divided into embryonic origin and postnatal origin.<span><sup>1</sup></span> Embryonic-origin macrophages are derived from the yolk sac and foetal liver during embryogenesis. They migrate to tissues early in development and maintain their populations with self-renewal mechanisms throughout the lifecycle. These macrophages are commonly found in tissues such as the brain (microglia), liver (Kupffer cells) and lungs (alveolar macrophages). In contrast, postnatal-origin macrophages are derived from hematopoietic stem cells in the bone marrow. During instances of injury, inflammation or tumorigenesis, monocytes are mobilized from the bone marrow to the blood and differentiate into macrophages upon reaching distinct tissues. Functionally, macrophages have traditionally been classified into M1 and M2 types based on in vitro activation modes. M1 macrophages exhibit pro-inflammatory properties, while M2 macrophages are involved in anti-inflammatory responses and tissue repair. M2 macrophages are further classified into subtypes M2a, M2b, M2c and M2d based on distinct activation signals.<span><sup>2</sup></span> However, recent perspectives suggest that macrophage functions exist on a spectrum rather than binary states, with significant heterogeneity across organs and diseases, challenging the simplistic polarization classification in explaining diverse pathophysiological processes.</p><p>The advent of single-cell RNA sequencing (scRNA-seq) technology has revolutionized the study of macrophages, providing unprecedented resolution to unravel their origin, heterogeneity and functional diversity in various physiological and pathological contexts. The integrated multi-organ scRNA-seq studies of macrophages aim to address the following key scientific questions: 1. Deciphering macrophage heterogeneity: Several studies integrated macrophages from normal tissues and tumour tissues of different organs for fine annotation, revealing significant heterogeneity of macrophages, rather than polarized classification.<span><sup>3, 4</sup></span> 2. Functional regulation of macrophage: In-depth analysis of macrophage function is crucial, such as inflammatory response and metabolism, thus informing therapeutic strategies aimed at modulating macrophage activity. For instance, TREM2+ macrophages have been identified in the brain, fat, liver, blood vessels and other physiological and pathological conditions, and have diverse functions such as phagocytosis, metabolism and pro-inflammatory effects.<span><sup>5</sup></span> SPP1+ macrophages have been identified in different diseases such as tumours and atrial fibrillation, and have diverse functions such as promoting fibrosis and regulating immunity.<span><sup>6</sup></span> 3. Inference of macrophage lineage origin and differentiation trajectories: Algorithms such as pseudotime and RNA velocity analysis are used to infer the lineage origin and differentiation trajectories of macrophages. Multi-organ single-cell RNA sequencing indicated that resident Timd4+Lyve1+Folr2+ macrophages originated from both yolk sac and foetal monocyte precursors<span><sup>7</sup></span> 4. Cell communication and cellular networks: Exploring the interaction of macrophages in parenchymal cells, interstitial cells and the microenvironment is at the forefront of research, also the difficulties in the field. Algorithms such as ligand−receptor analysis are used to infer the interaction.<span><sup>8</sup></span> (See Figure 1) However, these findings and inferences based on omics data require cautious interpretation and thorough validation, ranging from expression validation to functional validation. Techniques such as smFISH and immunostaining can verify the presence of subpopulations; lineage tracing animal models can validate the lineage origin and subtype transitions of macrophages; and functional studies can demonstrate the regulatory roles of specific molecules and cell−cell interactions.<span><sup>9</sup></span> (See Figure 1) Additionally, the integration of multiple omics approaches can significantly expand our understanding of macrophages. For example, spatial omics can pinpoint the spatial niches of macrophages and confirm their microenvironment and interactions in a spatial context. The integration of single-cell technologies such as scATAC-seq and CITE-seq can study different layers of systems biology, including the roles of epigenetics, transcription and proteins in physiological and pathological processes.<span><sup>9</sup></span></p><p>Currently, single-cell atlases of various organs in normal and disease states have been constructed, with substantial data available from Human Cell Atlas and Genotype-Tissue Expression for different organs and diseases. In cancer research, pan-cancer studies have integrated various data to finely annotate macrophages.<span><sup>3, 4</sup></span> However, integrated single-cell studies for specific organs and diseases are relatively few. Daccache et al. integrated published single-cell data from liver, kidney and lung, further clustering and annotating myeloid cells.<span><sup>10</sup></span> Significantly, an important metallothionein-expressing MAC_MT subpopulation was discovered in all three organs. Differentially expressed gene analysis and signature scoring confirmed the specificity of this subpopulation, which was further confirmed by additional dataset and immunostaining. To identify the potential biological significance of MAC_MT, authors performed ligand−receptor interaction analysis, revealing its role in cellular migration, vascularization and angiogenesis.</p><p>There are some advantages of such integrating single-cell research: 1. Large sample size and multi-organ integration: This approach mitigates the limitations of individual studies of low sample size and sample accessibility and enhances the statistical reliability. 2. Discovery of new subpopulations: The integration of data from various states and diseases facilitates the discovery of novel cell subpopulations. 3. Fine experimental design: It allows for fine experimental designs to address specific scientific questions, including gender and age differences, different regions of the same organ and temporal changes in disease states. 4. Integrate data from different species to explore the conservation of macrophage and increase translational significance. (See Figure 1) However, challenges remain in this field. 1. Varying data quality: The quality of single-cell data varies across studies, necessitating the careful selection of high-quality data and stringent quality control. 2. Batch effect handling and integration methods: Different studies exhibit batch effects that require thorough removal; however, excessive correction may lead to the loss of macrophage heterogeneity. 3. Differences in single-cell and single-nucleus analysis: Single-nucleus sequencing (snRNA-seq) is frequently employed in the examination of the heart, adipose tissue, kidneys and nervous system. However, it is essential to take into account the variations in transcript types when integrating the scRNA-seq and snRNA-seq data. scRNA-seq can detect cytoplasmic and nuclear transcripts, while snRNA-seq can only detect nuclear transcripts, which results in different detection abilities for macrophage surface marker genes and variations in profiling single-cell molecular phenotypes.<span><sup>9</sup></span></p><p>In the future, more research can depict single-cell landscapes of various tissues and diseases, aiming to discover tissue-specific and disease-specific macrophage subpopulations, thereby deepening our understanding of macrophages and aiding in the discovery of precise targeted therapies.</p><p>Xinjie Xu and Liang Chen contributed to conception and manuscript design. Xinjie Xu, Zichen Wu and draftedthe manuscript. Xinjie Xu and Zichen Wu prepared the figure. Xinjie Xu, Zhiwei Zeng, Jiaying Cao, Liang Chen participated in the revision of the manuscript. Xinjie Xu, Liang Chen were involved in funding acquisition. Allauthors read and approved the final manuscript.</p><p>The authors declare no conflict of interest.</p><p>The study was supported by the National Natural Science Foundation of China (82100377, 82470373, 823B2006); Beijing Nova Program (Z211100002121046); and National High Level Hospital Clinical Research Funding (2023-GSP-RC-01, 2023-GSP-ZD-2).</p><p>Not applicable.</p>","PeriodicalId":72605,"journal":{"name":"Clinical and translational discovery","volume":"4 5","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ctd2.70005","citationCount":"0","resultStr":"{\"title\":\"Integrating single-cell RNA sequencing data to decipher heterogeneity and function of macrophages in various organs and diseases\",\"authors\":\"Xinjie Xu, Zichen Wu, Zhiwei Zeng, Jiaying Cao, Liang Chen\",\"doi\":\"10.1002/ctd2.70005\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Macrophages are essential components of the innate immune system, playing crucial roles in host defence, immune surveillance and tissue repair processes. Developmentally, macrophages can be divided into embryonic origin and postnatal origin.<span><sup>1</sup></span> Embryonic-origin macrophages are derived from the yolk sac and foetal liver during embryogenesis. They migrate to tissues early in development and maintain their populations with self-renewal mechanisms throughout the lifecycle. These macrophages are commonly found in tissues such as the brain (microglia), liver (Kupffer cells) and lungs (alveolar macrophages). In contrast, postnatal-origin macrophages are derived from hematopoietic stem cells in the bone marrow. During instances of injury, inflammation or tumorigenesis, monocytes are mobilized from the bone marrow to the blood and differentiate into macrophages upon reaching distinct tissues. Functionally, macrophages have traditionally been classified into M1 and M2 types based on in vitro activation modes. M1 macrophages exhibit pro-inflammatory properties, while M2 macrophages are involved in anti-inflammatory responses and tissue repair. M2 macrophages are further classified into subtypes M2a, M2b, M2c and M2d based on distinct activation signals.<span><sup>2</sup></span> However, recent perspectives suggest that macrophage functions exist on a spectrum rather than binary states, with significant heterogeneity across organs and diseases, challenging the simplistic polarization classification in explaining diverse pathophysiological processes.</p><p>The advent of single-cell RNA sequencing (scRNA-seq) technology has revolutionized the study of macrophages, providing unprecedented resolution to unravel their origin, heterogeneity and functional diversity in various physiological and pathological contexts. The integrated multi-organ scRNA-seq studies of macrophages aim to address the following key scientific questions: 1. Deciphering macrophage heterogeneity: Several studies integrated macrophages from normal tissues and tumour tissues of different organs for fine annotation, revealing significant heterogeneity of macrophages, rather than polarized classification.<span><sup>3, 4</sup></span> 2. Functional regulation of macrophage: In-depth analysis of macrophage function is crucial, such as inflammatory response and metabolism, thus informing therapeutic strategies aimed at modulating macrophage activity. For instance, TREM2+ macrophages have been identified in the brain, fat, liver, blood vessels and other physiological and pathological conditions, and have diverse functions such as phagocytosis, metabolism and pro-inflammatory effects.<span><sup>5</sup></span> SPP1+ macrophages have been identified in different diseases such as tumours and atrial fibrillation, and have diverse functions such as promoting fibrosis and regulating immunity.<span><sup>6</sup></span> 3. Inference of macrophage lineage origin and differentiation trajectories: Algorithms such as pseudotime and RNA velocity analysis are used to infer the lineage origin and differentiation trajectories of macrophages. Multi-organ single-cell RNA sequencing indicated that resident Timd4+Lyve1+Folr2+ macrophages originated from both yolk sac and foetal monocyte precursors<span><sup>7</sup></span> 4. Cell communication and cellular networks: Exploring the interaction of macrophages in parenchymal cells, interstitial cells and the microenvironment is at the forefront of research, also the difficulties in the field. Algorithms such as ligand−receptor analysis are used to infer the interaction.<span><sup>8</sup></span> (See Figure 1) However, these findings and inferences based on omics data require cautious interpretation and thorough validation, ranging from expression validation to functional validation. Techniques such as smFISH and immunostaining can verify the presence of subpopulations; lineage tracing animal models can validate the lineage origin and subtype transitions of macrophages; and functional studies can demonstrate the regulatory roles of specific molecules and cell−cell interactions.<span><sup>9</sup></span> (See Figure 1) Additionally, the integration of multiple omics approaches can significantly expand our understanding of macrophages. For example, spatial omics can pinpoint the spatial niches of macrophages and confirm their microenvironment and interactions in a spatial context. The integration of single-cell technologies such as scATAC-seq and CITE-seq can study different layers of systems biology, including the roles of epigenetics, transcription and proteins in physiological and pathological processes.<span><sup>9</sup></span></p><p>Currently, single-cell atlases of various organs in normal and disease states have been constructed, with substantial data available from Human Cell Atlas and Genotype-Tissue Expression for different organs and diseases. In cancer research, pan-cancer studies have integrated various data to finely annotate macrophages.<span><sup>3, 4</sup></span> However, integrated single-cell studies for specific organs and diseases are relatively few. Daccache et al. integrated published single-cell data from liver, kidney and lung, further clustering and annotating myeloid cells.<span><sup>10</sup></span> Significantly, an important metallothionein-expressing MAC_MT subpopulation was discovered in all three organs. Differentially expressed gene analysis and signature scoring confirmed the specificity of this subpopulation, which was further confirmed by additional dataset and immunostaining. To identify the potential biological significance of MAC_MT, authors performed ligand−receptor interaction analysis, revealing its role in cellular migration, vascularization and angiogenesis.</p><p>There are some advantages of such integrating single-cell research: 1. Large sample size and multi-organ integration: This approach mitigates the limitations of individual studies of low sample size and sample accessibility and enhances the statistical reliability. 2. Discovery of new subpopulations: The integration of data from various states and diseases facilitates the discovery of novel cell subpopulations. 3. Fine experimental design: It allows for fine experimental designs to address specific scientific questions, including gender and age differences, different regions of the same organ and temporal changes in disease states. 4. Integrate data from different species to explore the conservation of macrophage and increase translational significance. (See Figure 1) However, challenges remain in this field. 1. Varying data quality: The quality of single-cell data varies across studies, necessitating the careful selection of high-quality data and stringent quality control. 2. Batch effect handling and integration methods: Different studies exhibit batch effects that require thorough removal; however, excessive correction may lead to the loss of macrophage heterogeneity. 3. Differences in single-cell and single-nucleus analysis: Single-nucleus sequencing (snRNA-seq) is frequently employed in the examination of the heart, adipose tissue, kidneys and nervous system. However, it is essential to take into account the variations in transcript types when integrating the scRNA-seq and snRNA-seq data. scRNA-seq can detect cytoplasmic and nuclear transcripts, while snRNA-seq can only detect nuclear transcripts, which results in different detection abilities for macrophage surface marker genes and variations in profiling single-cell molecular phenotypes.<span><sup>9</sup></span></p><p>In the future, more research can depict single-cell landscapes of various tissues and diseases, aiming to discover tissue-specific and disease-specific macrophage subpopulations, thereby deepening our understanding of macrophages and aiding in the discovery of precise targeted therapies.</p><p>Xinjie Xu and Liang Chen contributed to conception and manuscript design. Xinjie Xu, Zichen Wu and draftedthe manuscript. Xinjie Xu and Zichen Wu prepared the figure. Xinjie Xu, Zhiwei Zeng, Jiaying Cao, Liang Chen participated in the revision of the manuscript. Xinjie Xu, Liang Chen were involved in funding acquisition. Allauthors read and approved the final manuscript.</p><p>The authors declare no conflict of interest.</p><p>The study was supported by the National Natural Science Foundation of China (82100377, 82470373, 823B2006); Beijing Nova Program (Z211100002121046); and National High Level Hospital Clinical Research Funding (2023-GSP-RC-01, 2023-GSP-ZD-2).</p><p>Not applicable.</p>\",\"PeriodicalId\":72605,\"journal\":{\"name\":\"Clinical and translational discovery\",\"volume\":\"4 5\",\"pages\":\"\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2024-10-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ctd2.70005\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical and translational discovery\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ctd2.70005\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical and translational discovery","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ctd2.70005","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Integrating single-cell RNA sequencing data to decipher heterogeneity and function of macrophages in various organs and diseases
Macrophages are essential components of the innate immune system, playing crucial roles in host defence, immune surveillance and tissue repair processes. Developmentally, macrophages can be divided into embryonic origin and postnatal origin.1 Embryonic-origin macrophages are derived from the yolk sac and foetal liver during embryogenesis. They migrate to tissues early in development and maintain their populations with self-renewal mechanisms throughout the lifecycle. These macrophages are commonly found in tissues such as the brain (microglia), liver (Kupffer cells) and lungs (alveolar macrophages). In contrast, postnatal-origin macrophages are derived from hematopoietic stem cells in the bone marrow. During instances of injury, inflammation or tumorigenesis, monocytes are mobilized from the bone marrow to the blood and differentiate into macrophages upon reaching distinct tissues. Functionally, macrophages have traditionally been classified into M1 and M2 types based on in vitro activation modes. M1 macrophages exhibit pro-inflammatory properties, while M2 macrophages are involved in anti-inflammatory responses and tissue repair. M2 macrophages are further classified into subtypes M2a, M2b, M2c and M2d based on distinct activation signals.2 However, recent perspectives suggest that macrophage functions exist on a spectrum rather than binary states, with significant heterogeneity across organs and diseases, challenging the simplistic polarization classification in explaining diverse pathophysiological processes.
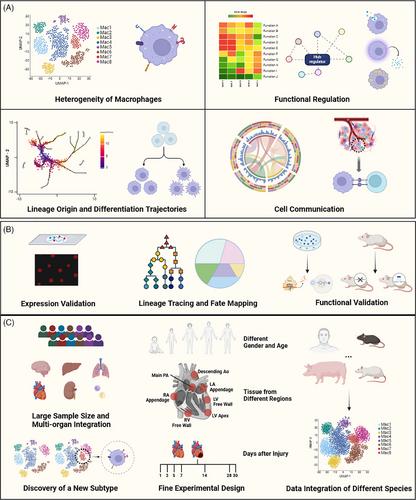
The advent of single-cell RNA sequencing (scRNA-seq) technology has revolutionized the study of macrophages, providing unprecedented resolution to unravel their origin, heterogeneity and functional diversity in various physiological and pathological contexts. The integrated multi-organ scRNA-seq studies of macrophages aim to address the following key scientific questions: 1. Deciphering macrophage heterogeneity: Several studies integrated macrophages from normal tissues and tumour tissues of different organs for fine annotation, revealing significant heterogeneity of macrophages, rather than polarized classification.3, 4 2. Functional regulation of macrophage: In-depth analysis of macrophage function is crucial, such as inflammatory response and metabolism, thus informing therapeutic strategies aimed at modulating macrophage activity. For instance, TREM2+ macrophages have been identified in the brain, fat, liver, blood vessels and other physiological and pathological conditions, and have diverse functions such as phagocytosis, metabolism and pro-inflammatory effects.5 SPP1+ macrophages have been identified in different diseases such as tumours and atrial fibrillation, and have diverse functions such as promoting fibrosis and regulating immunity.6 3. Inference of macrophage lineage origin and differentiation trajectories: Algorithms such as pseudotime and RNA velocity analysis are used to infer the lineage origin and differentiation trajectories of macrophages. Multi-organ single-cell RNA sequencing indicated that resident Timd4+Lyve1+Folr2+ macrophages originated from both yolk sac and foetal monocyte precursors7 4. Cell communication and cellular networks: Exploring the interaction of macrophages in parenchymal cells, interstitial cells and the microenvironment is at the forefront of research, also the difficulties in the field. Algorithms such as ligand−receptor analysis are used to infer the interaction.8 (See Figure 1) However, these findings and inferences based on omics data require cautious interpretation and thorough validation, ranging from expression validation to functional validation. Techniques such as smFISH and immunostaining can verify the presence of subpopulations; lineage tracing animal models can validate the lineage origin and subtype transitions of macrophages; and functional studies can demonstrate the regulatory roles of specific molecules and cell−cell interactions.9 (See Figure 1) Additionally, the integration of multiple omics approaches can significantly expand our understanding of macrophages. For example, spatial omics can pinpoint the spatial niches of macrophages and confirm their microenvironment and interactions in a spatial context. The integration of single-cell technologies such as scATAC-seq and CITE-seq can study different layers of systems biology, including the roles of epigenetics, transcription and proteins in physiological and pathological processes.9
Currently, single-cell atlases of various organs in normal and disease states have been constructed, with substantial data available from Human Cell Atlas and Genotype-Tissue Expression for different organs and diseases. In cancer research, pan-cancer studies have integrated various data to finely annotate macrophages.3, 4 However, integrated single-cell studies for specific organs and diseases are relatively few. Daccache et al. integrated published single-cell data from liver, kidney and lung, further clustering and annotating myeloid cells.10 Significantly, an important metallothionein-expressing MAC_MT subpopulation was discovered in all three organs. Differentially expressed gene analysis and signature scoring confirmed the specificity of this subpopulation, which was further confirmed by additional dataset and immunostaining. To identify the potential biological significance of MAC_MT, authors performed ligand−receptor interaction analysis, revealing its role in cellular migration, vascularization and angiogenesis.
There are some advantages of such integrating single-cell research: 1. Large sample size and multi-organ integration: This approach mitigates the limitations of individual studies of low sample size and sample accessibility and enhances the statistical reliability. 2. Discovery of new subpopulations: The integration of data from various states and diseases facilitates the discovery of novel cell subpopulations. 3. Fine experimental design: It allows for fine experimental designs to address specific scientific questions, including gender and age differences, different regions of the same organ and temporal changes in disease states. 4. Integrate data from different species to explore the conservation of macrophage and increase translational significance. (See Figure 1) However, challenges remain in this field. 1. Varying data quality: The quality of single-cell data varies across studies, necessitating the careful selection of high-quality data and stringent quality control. 2. Batch effect handling and integration methods: Different studies exhibit batch effects that require thorough removal; however, excessive correction may lead to the loss of macrophage heterogeneity. 3. Differences in single-cell and single-nucleus analysis: Single-nucleus sequencing (snRNA-seq) is frequently employed in the examination of the heart, adipose tissue, kidneys and nervous system. However, it is essential to take into account the variations in transcript types when integrating the scRNA-seq and snRNA-seq data. scRNA-seq can detect cytoplasmic and nuclear transcripts, while snRNA-seq can only detect nuclear transcripts, which results in different detection abilities for macrophage surface marker genes and variations in profiling single-cell molecular phenotypes.9
In the future, more research can depict single-cell landscapes of various tissues and diseases, aiming to discover tissue-specific and disease-specific macrophage subpopulations, thereby deepening our understanding of macrophages and aiding in the discovery of precise targeted therapies.
Xinjie Xu and Liang Chen contributed to conception and manuscript design. Xinjie Xu, Zichen Wu and draftedthe manuscript. Xinjie Xu and Zichen Wu prepared the figure. Xinjie Xu, Zhiwei Zeng, Jiaying Cao, Liang Chen participated in the revision of the manuscript. Xinjie Xu, Liang Chen were involved in funding acquisition. Allauthors read and approved the final manuscript.
The authors declare no conflict of interest.
The study was supported by the National Natural Science Foundation of China (82100377, 82470373, 823B2006); Beijing Nova Program (Z211100002121046); and National High Level Hospital Clinical Research Funding (2023-GSP-RC-01, 2023-GSP-ZD-2).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


