{"title":"了解前列腺癌多基因风险评分和差异的生物学基础:综合基因组分析","authors":"Wensheng Zhang, Kun Zhang","doi":"10.1177/11769351241276319","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>For prostate cancer (PCa), hundreds of risk variants have been identified. It remains unknown whether the polygenic risk score (PRS) that combines the effects of these variants is also a sufficiently informative metric with relevance to the molecular mechanisms of carcinogenesis in prostate. We aimed to understand the biological basis of PRS and racial disparities in the cancer.</p><p><strong>Methods: </strong>We performed a comprehensive analysis of the data generated (deposited in) by several genomic and/or transcriptomic projects (databases), including the GTEx, TCGA, 1000 Genomes, GEO and dbGap. PRS was constructed from 260 PCa risk variants that were identified by a recent trans-ancestry meta-analysis and contained in the GTEx dataset. The dosages of risk variants and the multi-ancestry effects on PCa incidence estimated by the meta-analysis were used in calculating individual PRS values.</p><p><strong>Results: </strong>The following novel results were obtained from our analyses. (1) In normal prostate samples from healthy European Americans (EAs), the expression levels of 540 genes (termed PRS genes) were associated with the PRS (<i>P</i> < .01). (2) Ubiquitin-proteasome system in high-PRS individuals' prostates was more active than that in low-PRS individuals' prostates. (3) Nine PRS genes play roles in the cancer progression-relevant parts, which are frequently hit by somatic mutations in PCa, of PI3K-Akt/RAS-MAPK/mTOR signaling pathways. (4) The expression profiles of the top significant PRS genes in tumor samples were capable of predicting malignant PCa relapse after prostatectomy. (5) The transcriptomic differences between African American and EA samples were incompatible with the patterns of the aforementioned associations between PRS and gene expression levels.</p><p><strong>Conclusions: </strong>This study provided unique insights into the relationship between PRS and the molecular mechanisms of carcinogenesis in prostate. The new findings, alongside the moderate but significant heritability of PCa susceptibility contributed by the risk variants, suggest the aptness and inaptness of PRS for explaining PCa and disparities.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"23 ","pages":"11769351241276319"},"PeriodicalIF":2.5000,"publicationDate":"2024-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11497523/pdf/","citationCount":"0","resultStr":"{\"title\":\"Understanding the Biological Basis of Polygenic Risk Scores and Disparities in Prostate Cancer: A Comprehensive Genomic Analysis.\",\"authors\":\"Wensheng Zhang, Kun Zhang\",\"doi\":\"10.1177/11769351241276319\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objectives: </strong>For prostate cancer (PCa), hundreds of risk variants have been identified. It remains unknown whether the polygenic risk score (PRS) that combines the effects of these variants is also a sufficiently informative metric with relevance to the molecular mechanisms of carcinogenesis in prostate. We aimed to understand the biological basis of PRS and racial disparities in the cancer.</p><p><strong>Methods: </strong>We performed a comprehensive analysis of the data generated (deposited in) by several genomic and/or transcriptomic projects (databases), including the GTEx, TCGA, 1000 Genomes, GEO and dbGap. PRS was constructed from 260 PCa risk variants that were identified by a recent trans-ancestry meta-analysis and contained in the GTEx dataset. The dosages of risk variants and the multi-ancestry effects on PCa incidence estimated by the meta-analysis were used in calculating individual PRS values.</p><p><strong>Results: </strong>The following novel results were obtained from our analyses. (1) In normal prostate samples from healthy European Americans (EAs), the expression levels of 540 genes (termed PRS genes) were associated with the PRS (<i>P</i> < .01). (2) Ubiquitin-proteasome system in high-PRS individuals' prostates was more active than that in low-PRS individuals' prostates. (3) Nine PRS genes play roles in the cancer progression-relevant parts, which are frequently hit by somatic mutations in PCa, of PI3K-Akt/RAS-MAPK/mTOR signaling pathways. (4) The expression profiles of the top significant PRS genes in tumor samples were capable of predicting malignant PCa relapse after prostatectomy. (5) The transcriptomic differences between African American and EA samples were incompatible with the patterns of the aforementioned associations between PRS and gene expression levels.</p><p><strong>Conclusions: </strong>This study provided unique insights into the relationship between PRS and the molecular mechanisms of carcinogenesis in prostate. The new findings, alongside the moderate but significant heritability of PCa susceptibility contributed by the risk variants, suggest the aptness and inaptness of PRS for explaining PCa and disparities.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"23 \",\"pages\":\"11769351241276319\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-10-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11497523/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351241276319\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351241276319","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Understanding the Biological Basis of Polygenic Risk Scores and Disparities in Prostate Cancer: A Comprehensive Genomic Analysis.
Objectives: For prostate cancer (PCa), hundreds of risk variants have been identified. It remains unknown whether the polygenic risk score (PRS) that combines the effects of these variants is also a sufficiently informative metric with relevance to the molecular mechanisms of carcinogenesis in prostate. We aimed to understand the biological basis of PRS and racial disparities in the cancer.
Methods: We performed a comprehensive analysis of the data generated (deposited in) by several genomic and/or transcriptomic projects (databases), including the GTEx, TCGA, 1000 Genomes, GEO and dbGap. PRS was constructed from 260 PCa risk variants that were identified by a recent trans-ancestry meta-analysis and contained in the GTEx dataset. The dosages of risk variants and the multi-ancestry effects on PCa incidence estimated by the meta-analysis were used in calculating individual PRS values.
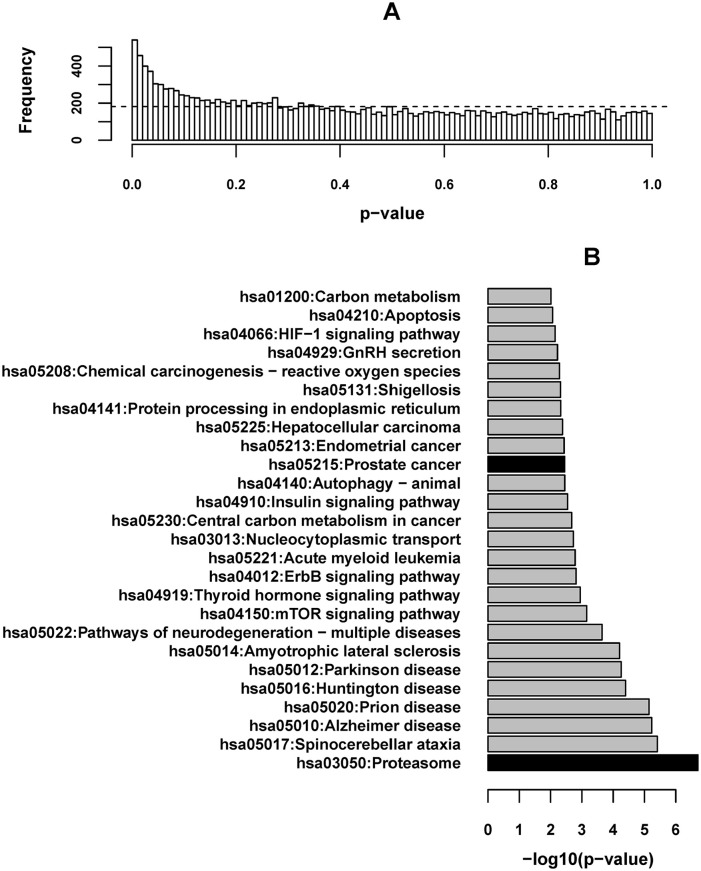
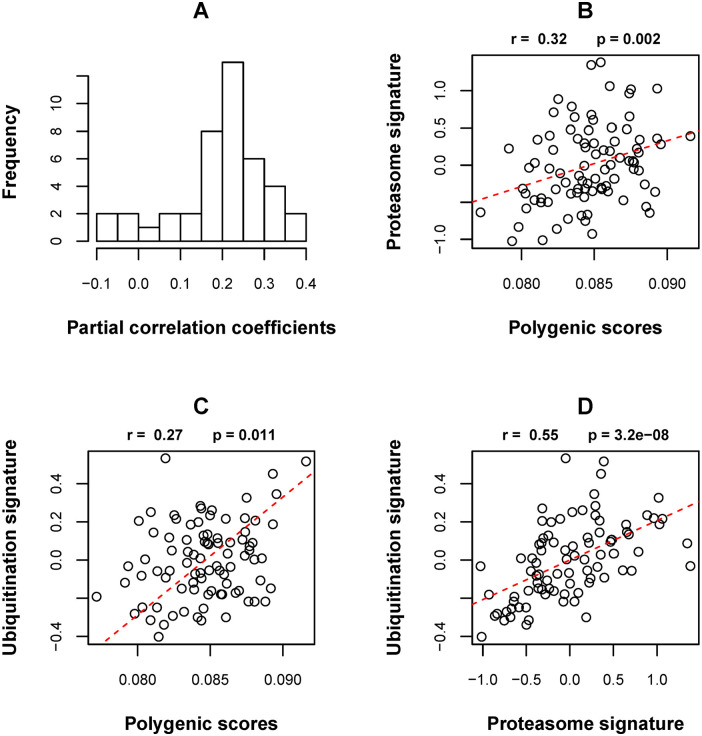
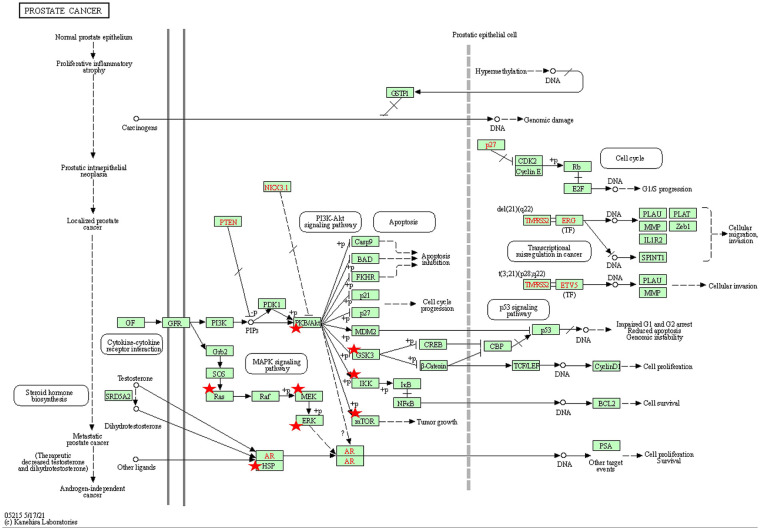
Results: The following novel results were obtained from our analyses. (1) In normal prostate samples from healthy European Americans (EAs), the expression levels of 540 genes (termed PRS genes) were associated with the PRS (P < .01). (2) Ubiquitin-proteasome system in high-PRS individuals' prostates was more active than that in low-PRS individuals' prostates. (3) Nine PRS genes play roles in the cancer progression-relevant parts, which are frequently hit by somatic mutations in PCa, of PI3K-Akt/RAS-MAPK/mTOR signaling pathways. (4) The expression profiles of the top significant PRS genes in tumor samples were capable of predicting malignant PCa relapse after prostatectomy. (5) The transcriptomic differences between African American and EA samples were incompatible with the patterns of the aforementioned associations between PRS and gene expression levels.
Conclusions: This study provided unique insights into the relationship between PRS and the molecular mechanisms of carcinogenesis in prostate. The new findings, alongside the moderate but significant heritability of PCa susceptibility contributed by the risk variants, suggest the aptness and inaptness of PRS for explaining PCa and disparities.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


